


畜牧兽医学报 ›› 2025, Vol. 56 ›› Issue (12): 6386-6396.doi: 10.11843/j.issn.0366-6964.2025.12.040
刘潇雨1( ), 李勇1(), 邱启官2, 程天印1, 段德勇1,*()
), 李勇1(), 邱启官2, 程天印1, 段德勇1,*()
收稿日期:2024-12-02
出版日期:2025-12-23
发布日期:2025-12-24
通讯作者:
段德勇
E-mail:a542640081@126.com;leo990522@163.com;kakayuan0980@163.com
作者简介:刘潇雨(2000-),男,湖南长沙人,硕士生,主要从事兽医寄生虫学研究,E-mail: a542640081@126.com刘潇雨与李勇为同等贡献作者
基金资助:
LIU Xiaoyu1(), LI Yong1(), QIU Qiguan2, CHENG Tianyin1, DUAN Deyong1,*()
Received:2024-12-02
Online:2025-12-23
Published:2025-12-24
Contact:
DUAN Deyong
E-mail:a542640081@126.com;leo990522@163.com;kakayuan0980@163.com
摘要:
旨在探究孟加拉虎体表不同饱血状态长角血蜱雌蜱中肠菌群结构的特点。本研究采集孟加拉虎体表饱血(fully engorged,FF)与半饱血(half engorged,HF)长角血蜱雌蜱,在无菌条件下收集各组蜱中肠内容物,提取各组样本细菌总DNA,PCR扩增16S rRNA V3-V4区,构建文库,Illumina NovaSeq平台双末端测序,测序数据经拼接、过滤、降噪后,进行Amplicon Sequence Variant(ASV)和多样性分析。结果显示,FF、HF获得的有效序列分别为100 021条和82 657条,聚类后FF、HF分别获得ASVs为259、541个,两组样品共有的ASVs为116个。在门水平上,两组样本均以变形菌门、厚壁菌门、拟杆菌门、放线菌门为优势菌门;变形菌门在FF中的含量大于HF,其它三个优势菌门反之。在属水平上,柯克斯氏体属、Muribaculaceae、棒状杆菌属、代尔夫特菌属、葡萄球菌属为FF和HF中的共有优势菌属,其中柯克斯氏体属在两组样本中含量均较高,且在FF中的含量高于HF;FF、HF中特有且相对丰度较高的菌属分别为Schlegelella、普雷沃氏菌属等。在种水平上,两组样本以柯克斯氏体属内共生体含量最高,其在FF和HF中的分布特点与柯克斯氏体属在两组样本中的分布特点相一致。综上,长角血蜱雌蜱中肠微生物菌群结构复杂,不同饱血状态雌蜱中肠菌群结构存在一定差异,且同一菌属相对丰度不同。
中图分类号:
刘潇雨, 李勇, 邱启官, 程天印, 段德勇. 孟加拉虎体表不同饱血状态长角血蜱中肠菌群结构的分析[J]. 畜牧兽医学报, 2025, 56(12): 6386-6396.
LIU Xiaoyu, LI Yong, QIU Qiguan, CHENG Tianyin, DUAN Deyong. Analysis of the Midgut Microflora of Different Engorged Haemaphysalis longicornis Collected from Panthera tigris tigris[J]. Acta Veterinaria et Zootechnica Sinica, 2025, 56(12): 6386-6396.

图 1
长角血蜱两组样品ASVs分布韦恩图与β多样性分析 a. ASVs分布韦恩图;b. 距离矩阵热图。HF.半饱血状态下长角血蜱中肠内容物;FF.饱血状态下长角血蜱中肠内容物,下同"
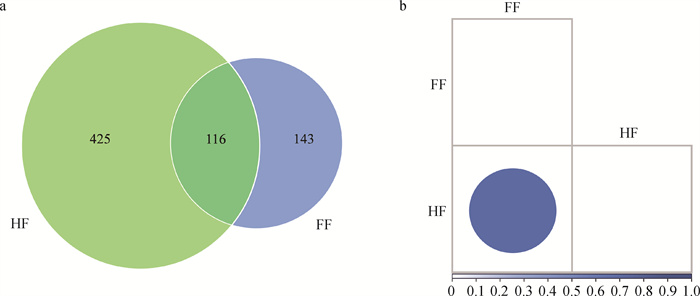
表 1
样品的细菌多样性指数"
| 样本 Sample | 测序覆盖率 Good’s coverage | Chao1多样性指数 Chao1 | 香农多样性指数 Shannon | 辛普森多样性指数 Simpson |
| HF | 1.000 | 541.000 | 3.737 | 0.635 |
| FF | 1.000 | 259.375 | 1.519 | 0.290 |
图 2
两组样本中16个细菌门相对丰度的分布特点"
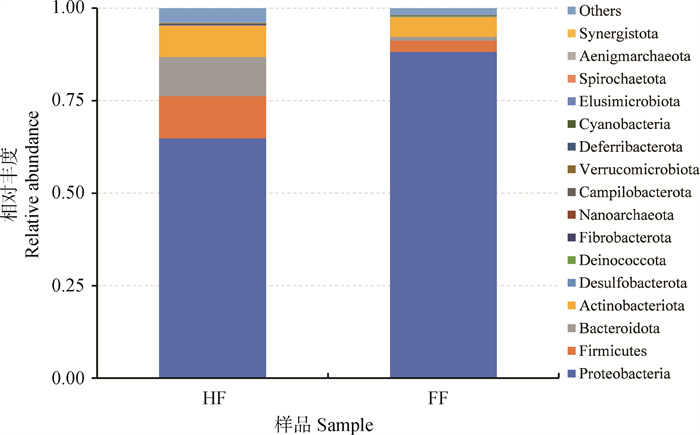
图 3
相对丰度前15的细菌属在两组样本中的分布特点"
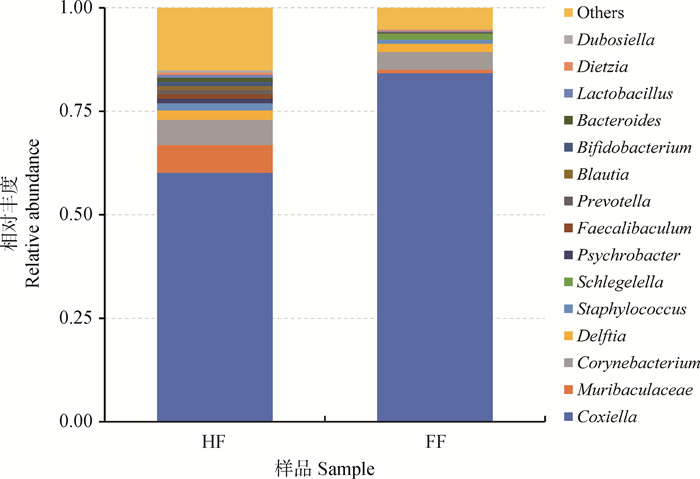
表 2
两组样本中前15个共有细菌属以外其他共有细菌属的相对丰度"
| 细菌属 Bacterial genus | 细菌属在每个样本中的相对丰度 Relative abundance of bacteria genus in each sample | |
| FF | HF | |
| 副拟杆菌属Parabacteroides | 0.000 026 508 | 0.004 718 482 |
| 罗姆布茨菌属Romboutsia | 0.001 192 875 | 0.004 387 128 |
| Lachnoclostridium | 0.000 039 762 | 0.00 331 354 |
| 梭状芽孢杆菌属Clostridium sensu stricto | 0.000 119 287 | 0.003 207 507 |
| 真杆菌属Eubacterium | 0.000 026 508 | 0.002 809 882 |
| 毛螺菌科NK4A136属Lachnospiraceae NK4A136 | 0.000 675 962 | 0.002 783 374 |
| Rikenellaceae RC9 gut group | 0.000 198 812 | 0.002 107 412 |
| 罗斯氏菌属Rothia | 0.002 054 395 | 0.000 384 371 |
| 短杆菌属Brevibacterium | 0.001 047 079 | 0.001 802 566 |
| 短状杆菌属Brachybacterium | 0.000 861 521 | 0.001 723 041 |
| 肠球菌属Enterococcus | 0.001 577 245 | 0.000 092 779 |
| Anaerotruncus | 0.000 543 421 | 0.00 149 772 |
| Incertae sedis | 0.000 119 287 | 0.001 259 145 |
| 螺杆菌属Helicobacter | 0.001 179 620 | 0.000 106 033 |
| 微球菌属Micrococcus | 0.000 344 608 | 0.001 153 112 |
| 苏黎世杆菌属Turicibacter | 0.00 015 905 | 0.000 980 808 |
| Dysgonomonas | 0.000 013 254 | 0.000 848 266 |
| 另枝菌属Alistipes | 0.000 556 675 | 0.000 622 946 |
| 阿克曼氏菌属Akkermansia | 0.000 013 254 | 0.000 583 183 |
| 无色杆菌属Achromobacter | 0.000 530 166 | 0.000 424 133 |
| Cutibacterium | 0.000 490 404 | 0.000 172 304 |
| Eubacterium xylanophilum group | 0.000 106 033 | 0.000 463 896 |
| 不动杆菌属Acinetobacter | 0.000 384 371 | 0.000 463 896 |
| A2 | 0.000 039 762 | 0.000 450 642 |
| GCA-900066575 | 0.000 039 762 | 0.000 437 387 |
| 毛螺菌科UCG-006属Lachnospiraceae UCG-006 | 0.000 079 525 | 0.000 397 625 |
| 溶杆菌属Lysobacter | 0.000 185 558 | 0.000 384 371 |
| Clostridia vadinBB60 group | 0.000 371 117 | 0.000 357 862 |
| 巨大球菌属Macrococcus | 0.000 053 017 | 0.0 003 181 |
| Lawsonella | 0.000 291 592 | 0.000 265 083 |
| 布兰汉氏菌属Alkanindiges | 0.000 039 762 | 0.000 291 592 |
| Colidextribacter | 0.000 092 779 | 0.000 238 575 |
| 栖水菌属Enhydrobacter | 0.000 092 779 | 0.000 225 321 |
| Deep Sea Euryarchaeotic Group | 0.000 119 287 | 0.000 092 779 |
| 食酸菌属Acidovorax | 0.000 026 508 | 0.000 106 033 |
| ASF356 | 0.000 053 017 | 0.000 106 033 |
| 考克氏菌属Kocuria | 0.000 039 762 | 0.000 092 779 |
表 3
HF中特有细菌种名称及相对丰度"
| 细菌种 Bacterial species | 相对丰度 Relative abundance | 细菌种 Bacterial species | 相对丰度 Relative abundance | |
| Clostridium aldenense | 0.001 219 383 | bacterium XPD3003 | 0.000 146 | |
| Blattella germanica | 0.001 153 112 | Marinococcus sp. | 0.000 133 | |
| 产琥珀酸丝状杆菌Fibrobacter succinogenes | 0.000 781 996 | Luteimonas sp. | 0.000 119 | |
| Dorea sp. | 0.000 649 454 | Chryseobacterium salipaludis | 0.000 119 | |
| 栖瘤胃普雷沃氏菌Prevotella ruminicola | 0.000 583 183 | bacterium enrichment | 0.000 106 | |
| Bacteroides sp. | 0.000 490 404 | Treponema bryantii | 0.000 106 | |
| bacterium AC2043 | 0.000 490 404 | bacterium YRD2003 | 0.000 106 | |
| rumen bacterium | 0.000 424 133 | Azospirillum sp. | 0.000 093 | |
| Fibrobacter sp. | 0.000 397 625 | bacterium ND2018 | 0.000 093 | |
| 梭菌目细菌Clostridiales bacterium | 0.000 344 608 | bacterium MD2012 | 0.000 066 | |
| Ruminococcus sp. | 0.000 291 592 | bacterium YGB2004 | 0.000 066 | |
| 黄化瘤胃球菌Ruminococcus flavefaciens | 0.000 278 337 | Chryseobacterium chengduensis | 0.000 066 | |
| 拟杆菌目细菌Bacteroidales bacterium | 0.000 278 337 | rumen bacterium | 0.000 066 | |
| Adlercreutzia mucosicola | 0.000 212 067 | bacterium MB2022 | 0.000 066 | |
| rumen bacterium | 0.000 185 558 | bacterium V9D2012 | 0.000 066 | |
| 汤氏不动杆菌Acinetobacter towneri | 0.000 172 304 | 布雷恩特普雷沃菌Prevotella bryantii | 0.000 066 | |
| bacterium V9D2002 | 0.00 015 905 | |||
| bacterium WCE2007 | 0.000 159 |
| 1 |
RAINEY T , OCCI J L , ROBBINS R G , et al. Discovery of Haemaphysalis longicornis (Ixodida: Ixodidae) parasitizing a sheep in New Jersey, United States[J]. J Med Entomol, 2018, 55 (3): 757- 759.
doi: 10.1093/jme/tjy006 |
| 2 |
ZHENG H , YU Z , ZHOU L , et al. Seasonal abundance and activity of the hard tick Haemaphysalis longicornis (Acari: Ixodidae) in North China[J]. Exp Appl Acarol, 2012, 56 (2): 133- 141.
doi: 10.1007/s10493-011-9505-x |
| 3 | 余增莹. 长角血蜱和嗜群血蜱ITS-2、COⅠ和COⅡ基因的序列分析研究[D]. 雅安: 四川农业大学, 2008. |
| YU Z Y. Molecular phylogenetics analysis of Haemaphysalis longicornis and H. conicinna based on ITS-2, COⅠ and COⅡgenes[D]. Ya'an: Sichuan Agricultural University, 2008. (in Chinese) | |
| 4 | 于东海. 辽宁省部分地区蜱种分布及携带主要病原的调查研究[D]. 沈阳: 沈阳农业大学, 2020. |
| YU D H. Investigation on the distribution of ticks and their main pathogens in some areas of Liaoning Province[D]. Shenyang: Shenyang Agricultural University, 2020. (in Chinese) | |
| 5 | 朱振勤, 陈季武, 夏明仪. 吸血期蓖麻硬蜱雌虫体内周身性莱姆病螺旋体生长动态的组织学研究[J]. 中国人兽共患病杂志, 1998, 14 (3): 16- 20. |
| ZHU Z Q , CHEN J W , XIA M Y . Growth kinetics of systemic lyme disease spirochetes in blood-feeding Ixodes ricinus females[J]. Chinese Journal of Zoonoses, 1998, 14 (3): 16- 20. | |
| 6 |
SMITH T A , DRISCOLL T , GILLESPIE J J , et al. A Coxiella-like endosymbiont is a potential vitamin source for the Lone Star tick[J]. Genome Biol Evol, 2015, 7 (3): 831- 838.
doi: 10.1093/gbe/evv016 |
| 7 | 胡永红, 刘敬泽. 长角血蜱成蜱中肠结构的比较研究[J]. 中国昆虫学会, 2004, 680- 682. |
| HU Y H , LIU J Z . A comparative study of the structure of the midgut of adult Haemaphysalis longicornis[J]. Entomological Society of China, 2004, 680- 682. | |
| 8 | 廖芷卉, 陈宏智, 唐昊, 等. 羊源长角血蜱吸血后中肠优势菌群分析[J]. 中国动物传染病学报, 2015, 23 (4): 61- 64. |
| LIAO Z H , CHEN H Z , TANG H , et al. Analysis of dominant midgut bacterial communities in Haemaphysalis longicornis from sheep after blood feeding[J]. Chinese Journal of Animal Infectious Diseases, 2015, 23 (4): 61- 64. | |
| 9 | 唐莲, 江叔芳, 唐欢. 狼源和羊源长角血蜱体内菌群分析[J]. 湖南畜牧兽医, 2016 (4): 36- 39. |
| TANG L , JIANG S F , TANG H . Analysis of the internal microflora of Haemaphysalis longicornis from wolf and sheep[J]. Hunan Animal science and Veterinary Medicine, 2016 (4): 36- 39. | |
| 10 |
CAO R , REN Q , LUO J , et al. Analysis of microorganism diversity in Haemaphysalis longicornis from Shaanxi, China, based on metagenomic sequencing[J]. Front Genet, 2021, 12, 723773.
doi: 10.3389/fgene.2021.723773 |
| 11 | 党利红, 张瑞玲. 长角血蜱雌雄个体间共生菌多样性比较[J]. 中国病原生物学杂志, 2018, 13 (5): 520- 523. |
| DANG L H , ZHANG R L . Comparison of the microbe diversity in Haemaphysalis longicornis males and females[J]. Journal of Pathogen Biology, 2018, 13 (5): 520- 523. | |
| 12 | MAGOC T , SALZBERG S L . FLASH: fast length adjustment of short reads to improve genome assemblies[J]. Bioinformatics (Oxford, England), 2011, 27 (21): 2957- 2963. |
| 13 |
HAAS B J , GEVERSD , EARL A M , et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons[J]. Genome Res, 2011, 21 (3): 494- 504.
doi: 10.1101/gr.112730.110 |
| 14 |
CALLAHAN B J , MCMURDIE P J , ROSEN M J , et al. DADA2: High-resolution sample inference from Illumina amplicon data[J]. Nat Methods, 2016, 13 (7): 581- 583.
doi: 10.1038/nmeth.3869 |
| 15 |
LI M , SHAO D , ZHOU J , et al. Signatures within esophageal microbiota with progression of esophageal squamous cell carcinoma[J]. Chin J Cancer Res, 2020, 32 (6): 755- 767.
doi: 10.21147/j.issn.1000-9604.2020.06.09 |
| 16 |
BOKULICH N A , KAEHLER B D , RIDEOUT J R , et al. Optimizing taxonomic classification of marker-gene amplicon sequences with QⅡME 2's q2-feature-classifier plugin[J]. Microbiome, 2018, 6 (1): 90.
doi: 10.1186/s40168-018-0470-z |
| 17 |
CALLAHAN B J , MCMURDIE P J , HOLMES S P . Exact sequence variants should replace operational taxonomic units in marker-gene data analysis[J]. ISME J, 2017, 11 (12): 2639- 2643.
doi: 10.1038/ismej.2017.119 |
| 18 |
LOZUPONE C , KNIGHT R . UniFrac: a new phylogenetic method for comparing microbial communities[J]. Appl Environ Microb, 2005, 71 (12): 8228- 8235.
doi: 10.1128/AEM.71.12.8228-8235.2005 |
| 19 |
OSIMANI A , MILANOVIC V , GAROFALO C , et al. Revealing the microbiota of marketed edible insects through PCR-DGGE, metagenomic sequencing and real-time PCR[J]. Int J Food Microbiol, 2018, 276, 54- 62.
doi: 10.1016/j.ijfoodmicro.2018.04.013 |
| 20 |
XU X L , CHENG T Y , YANG H , et al. Identification of intestinal bacterial flora in Rhipicephalus microplus ticks by conventional methods and PCR-DGGE analysis[J]. Exp Appl Acarol, 2015, 66 (2): 257- 268.
doi: 10.1007/s10493-015-9896-1 |
| 21 |
段德勇, 程天印. 不同饱血状态微小牛蜱中肠和唾液菌群结构的分析[J]. 畜牧兽医学报, 2017, 48 (3): 530- 537.
doi: 10.11843/j.issn.0366-6964.2017.03.017 |
|
DUAN D Y , CHENG T Y . Bacterial flora analysis of saliva and midgut contents from partially or fully engorged female adult Rhipicephalus microplus[J]. Acta Veterinaria et Zootechnica Sinica, 2017, 48 (3): 530- 537.
doi: 10.11843/j.issn.0366-6964.2017.03.017 |
|
| 22 |
段德勇, 周鸿铭, 程天印. 吸血行为对草原革蜱和森林革蜱中肠菌群结构的影响[J]. 畜牧兽医学报, 2020, 51 (1): 128- 136.
doi: 10.11843/j.issn.0366-6964.2020.01.015 |
|
DUAN D Y , ZHOU H M , CHENG T Y . Effects of blood-sucking behavior on the microbial community in the midgut of Dermacentor nuttalli and Dermacentor silvarum[J]. Acta Veterinaria et Zootechnica Sinica, 2020, 51 (1): 128- 136.
doi: 10.11843/j.issn.0366-6964.2020.01.015 |
|
| 23 |
HIRAI J , NAGAI S , HIDAKA K . Evaluation of metagenetic community analysis of planktonic copepods using Illumina MiSeq: Comparisons with morphological classification and metagenetic analysis using Roche 454[J]. PLoS One, 2017, 12 (7): e0181452.
doi: 10.1371/journal.pone.0181452 |
| 24 |
GUO Y , WANG R , ZHAO Y , et al. Study on the relationship between microbial composition and living environment in important medical mites based on Illumina MiSeq sequencing technology[J]. J Med Entomol, 2020, 57 (4): 1049- 1056.
doi: 10.1093/jme/tjaa034 |
| 25 |
CALLAHAN B J , WONG J , HEINER C , et al. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution[J]. Nucleic Acids Res, 2019, 47 (18): e103.
doi: 10.1093/nar/gkz569 |
| 26 | AMIR A , MCDONALD D , NAVAS-MOLINA J A , et al. Deblur rapidly resolves single-nucleotide community sequence patterns[J]. mSystems, 2017, 2 (2): e00191- 16. |
| 27 | 韩娜, 张琳, 张雯, 等. 长角血蜱细菌群落结构及多样性研究[J]. 中国媒介生物学及控制杂志, 2016, 27 (5): 426- 431. |
| HAN N , ZHANG L , ZHANG W , et al. Studies on the composition and diversity of the bacterial community in Haemaphysalis longicornis[J]. Chinese Journal of Vector Biology and Control, 2016, 27 (5): 426- 431. | |
| 28 |
LALZAR I , HARRUS S , MUMCUOGLU K Y , et al. Composition and seasonal variation of Rhipicephalus turanicus and Rhipicephalus sanguineus bacterial communities[J]. Appl Environ Microb, 2012, 78 (12): 4110- 4116.
doi: 10.1128/AEM.00323-12 |
| 29 | WANG M , ZHU D , DAI J , et al. Tissue localization and variation of major symbionts in Haemaphysalis longicornis, Rhipicephalus haemaphysaloides, and Dermacentor silvarum in China[J]. Appl Environ Microb, 2018, 84 (10): e00029- 18. |
| 30 |
KLYACHKO O , STEIN B D , GRINDLE N , et al. Localization and visualization of a Coxiella-type symbiont within the lone star tick, Amblyomma americanum[J]. Appl Environ Microb, 2007, 73 (20): 6584- 6594.
doi: 10.1128/AEM.00537-07 |
| 31 |
BUYSSE M , PLANTARD O , MCCOY K D , et al. Tissue localization of Coxiella-like endosymbionts in three European tick species through fluorescence in situ hybridization[J]. Ticks Tick Borne Dis, 2019, 10 (4): 798- 804.
doi: 10.1016/j.ttbdis.2019.03.014 |
| 32 |
BEN-YOSEF M , ROT A , MAHAGNA M , et al. Coxiella-like endosymbiont of Rhipicephalus sanguineus is required for physiological processes during ontogeny[J]. Front Microbiol, 2020, 11, 493.
doi: 10.3389/fmicb.2020.00493 |
| 33 |
ZHONG Z , ZHONG T , PENG Y , et al. Symbiont-regulated serotonin biosynthesis modulates tick feeding activity[J]. Cell Host Microbe, 2021, 29 (10): 1545- 1557.
doi: 10.1016/j.chom.2021.08.011 |
| 34 |
MIRA A , MORAN N A . Estimating population size and transmission bottlenecks in maternally transmitted endosymbiotic bacteria[J]. Microb Ecol, 2002, 44 (2): 137- 143.
doi: 10.1007/s00248-002-0012-9 |
| 35 |
CAMARGO C H , FERREIRA A M , JAVARONI E , et al. Microbiological characterization of Delftia acidovorans clinical isolates from patients in an intensive care unit in Brazil[J]. Diagn Micr Infec Dis, 2014, 80 (4): 330- 333.
doi: 10.1016/j.diagmicrobio.2014.09.001 |
| 36 | 万玉香, 刘云, 秦琴. 食酸代尔夫特菌感染性脑膜炎一例[J]. 海军医学杂志, 2018, 39 (6): 593- 594. |
| WAN Y X , LIU Y , QIN Q . A case of Delftia acidovorans infective meningitis[J]. Journal of Navy Medicine, 2018, 39 (6): 593- 594. | |
| 37 |
LAVIAD-SHITRIT S , SHARABY Y , SELA R , et al. Copper and chromium exposure affect chironomid larval microbiota composition[J]. Sci Total Environ, 2021, 771, 145330.
doi: 10.1016/j.scitotenv.2021.145330 |
| 38 |
SUN Z , LU Y , ZHANG H , et al. Effects of BmCPV infection on silkworm Bombyx mori intestinal bacteria[J]. PLoS One, 2016, 11 (1): e0146313.
doi: 10.1371/journal.pone.0146313 |
| 39 |
MEREGHETTI V , CHOUAIA B , LIMONTA L , et al. Evidence for a conserved microbiota across the different developmental stages of Plodia interpunctella[J]. Insect Sci, 2019, 26 (3): 466- 478.
doi: 10.1111/1744-7917.12551 |
| 40 |
PRIYA N G , OJHA A , KAJLA M K , et al. Host plant induced variation in gut bacteria of Helicoverpa armigera[J]. PLoS One, 2012, 7 (1): e30768.
doi: 10.1371/journal.pone.0030768 |
| 41 |
VEGA F E , EMCHE S , SHAO J , et al. Cultivation and genome sequencing of bacteria isolated from the coffee berry borer (Hypothenemus hampei), with emphasis on the role of caffeine degradation[J]. Front Microbiol, 2021, 12, 644768.
doi: 10.3389/fmicb.2021.644768 |
| 42 |
STRANO C P , MALACRINO A , CAMPOLO O , et al. Influence of host plant on Thaumetopoea pityocampa gut bacterial community[J]. Microb Ecol, 2018, 75 (2): 487- 494.
doi: 10.1007/s00248-017-1019-6 |
| 43 |
WANG Y , GILBREATH T M , KUKUTLA P , et al. Dynamic gut microbiome across life history of the malaria mosquito Anopheles gambiae in Kenya[J]. PLoS One, 2011, 6 (9): e24767.
doi: 10.1371/journal.pone.0024767 |
| 44 |
SHARMA G , GARG N , HASAN S , et al. An insight into its characteristics and associated virulence factors[J]. Microb Pathogenesis, 2022, 169, 105673.
doi: 10.1016/j.micpath.2022.105673 |
| 45 |
BERTELSEN A , ELBORN J S , SCHOCK B C . Microbial interaction: Prevotella spp. reduce P. aeruginosa induced inflammation in cystic fibrosis bronchial epithelial cells[J]. J Cyst Fibros, 2021, 20 (4): 682- 691.
doi: 10.1016/j.jcf.2021.04.012 |
| 46 | 张洁, 张力莉, 徐晓锋. 反刍动物瘤胃内普雷沃氏菌的研究进展[J]. 中国饲料, 2020 (7): 17- 21. |
| ZHANG J , ZHANG L L , XU X F . Research progress of Prevotella in the rumen of ruminants[J]. China Feed, 2020 (7): 17- 21. | |
| 47 |
TYAGI K , TYAGI I , KUMAR V . Insights into the gut bacterial communities of spider from wild with no evidence of phylosymbiosis[J]. Saudi J Biol Sci, 2021, 28 (10): 5913- 5924.
doi: 10.1016/j.sjbs.2021.06.059 |
| 48 |
OLIVEIRA T M P , SANABANI S S , SALLUM M A M . Bacterial diversity associated with the abdomens of naturally Plasmodium-infected and non-infected Nyssorhynchus darlingi[J]. BMC Microbiol, 2020, 20 (1): 180.
doi: 10.1186/s12866-020-01861-0 |
| 49 |
ZENG J Y , SHI J H , GUO J X , et al. Variation in the pH of experimental diets affects the performance of Lymantria dispar asiatica larvae and its gut microbiota[J]. Arch Insect Biochem Physiol, 2020, 103 (4): e21654.
doi: 10.1002/arch.21654 |
| 50 |
KALAPPA D M , SUBRAMANI P A , BASAVANNA S K , et al. Influence of midgut microbiota in Anopheles stephensi on Plasmodium berghei infections[J]. Malar J, 2018, 17 (1): 385.
doi: 10.1186/s12936-018-2535-7 |
| 51 | 乔林赏. 万古霉素鞘内阶梯浓度注射对颅内葡萄球菌属感染患者的临床疗效评价[J]. 抗感染药学, 2020, 17 (1): 139- 142. |
| QIAO L S . Evaluation of the clinical efficacy of intrathecal step concentration injection of vancomycin in patients with intracranial Staphylococcus infections[J]. Anti-Infection Pharmacy, 2020, 17 (1): 139- 142. | |
| 52 |
VALIENTE MORO C , THIOULOUSE J , CHAUVE C , et al. Bacterial taxa associated with the hematophagous mite Dermanyssus gallinae detected by 16S rRNA PCR amplification and TTGE fingerprinting[J]. Res Microbiol, 2009, 160 (1): 63- 70.
doi: 10.1016/j.resmic.2008.10.006 |
| 53 | SOUKUP P , VETROVDKY T , STIBLIK P , et al. Termites are associated with external species-specific bacterial communities[J]. Appl Environ Microbiol, 2021, 87 (2): e02042- 20. |
| 54 |
DALL'AGNOL B , MCCULLOCH J A , MAYER F Q , et al. Molecular characterization of bacterial communities of two neotropical tick species (Amblyomma aureolatum and Ornithodoros brasiliensis) using rDNA 16S sequencing[J]. Ticks Tick Borne Dis, 2021, 12 (5): 101746.
doi: 10.1016/j.ttbdis.2021.101746 |
| 55 | 李春红. 三种硬蜱肠道菌群及其部分菌株生物学特性分析[D]. 长沙: 湖南农业大学, 2012. |
| LI C H. Intesinal flora in three hard ticks and biological characteristics analysis of part strains[D]. Changsha: Hunan Agricultural University, 2012. (in Chinese) | |
| 56 | 向昱龙, 周敬祝, 刘英, 等. 贵州省部分地区蜱及其携带细菌调查[J]. 中国媒介生物学及控制杂志, 2022, 33 (1): 148- 152. |
| XIANG Y L , ZHOU J Z , LIU Y , et al. An investigation of ticks and tick-borne bacteria in some areas of Guizhou province, China[J]. Chinese Journal of Vector Biology and Control, 2022, 33 (1): 148- 152. | |
| 57 |
TUANUDOM R , YURAYART N , RODKHUM C , et al. Diversity of midgut microbiota in laboratory-colonized and field-collected Aedes albopictus (Diptera: Culicidae): A preliminary study[J]. Heliyon, 2021, 7 (10): e08259.
doi: 10.1016/j.heliyon.2021.e08259 |
| [1] | 杨文慧, 王菲菲, 李晨雷, 孙宇, 秦俊杰, 朱浩, 郭延生. 基于奶牛瘤胃菌群分析四君子散健脾效应的作用机制[J]. 畜牧兽医学报, 2025, 56(12): 6487-6501. |
| [2] | 娜梅拉, 李科南, 杜海东, 郭文亮, 娜仁花. 不同日龄内蒙古白绒山羊瘤胃及粪便真菌多样性差异研究[J]. 畜牧兽医学报, 2024, 55(8): 3526-3540. |
| [3] | 郑焕琴, 姜晓敏, 岳红, 王宝岩, 刘洋, 张兴晓, 张建龙, 朱洪伟. 猫1型疱疹病毒分离鉴定及部分生物学特性分析[J]. 畜牧兽医学报, 2024, 55(7): 3040-3048. |
| [4] | 李聪聪, 黄子珂, 黄念旎, 马诗语, 刘晗, 肖志标, 宋果, 蒋亮, 彭为波, 杨联熙, 郭云涛, 黄生强. 九疑山兔线粒体基因组组装及系统进化分析[J]. 畜牧兽医学报, 2024, 55(10): 4417-4427. |
| [5] | 付涵, 卢冲, 缪荣浩, 卢亚宾, 李建龙, 刘建华, 耿明阳, 郭庆勇, 买占海, 况玲. 流产对母马阴道和肠道菌群多样性的影响及阴道细菌的分离鉴定[J]. 畜牧兽医学报, 2024, 55(10): 4700-4719. |
| [6] | 张道亮, 丁红研, 王留幸, 邰文俊, 孔昊, 赵畅, 冯士彬, 王希春, 薛艳锋, 吴金节, 李玉. 瘤胃酸中毒对山羊胃肠道功能、形态和菌群的影响[J]. 畜牧兽医学报, 2024, 55(10): 4760-4772. |
| [7] | 吴祎程, 冉涛, 周传社, 谭支良. 宏基因组学技术分析山羊瘤胃病毒的多样性[J]. 畜牧兽医学报, 2023, 54(7): 2932-2941. |
| [8] | 李蔚, 张强, 瞿嘉豪, 吴亚平, 胡若辰, 贾若艺, 郭如海, 马清义, 潘广林, 王兴龙. 大熊猫肠道菌群年龄演替规律分析[J]. 畜牧兽医学报, 2023, 54(6): 2619-2630. |
| [9] | 秦蕾, 吴慧敏, 徐琦琦, 陈万昭, 王东, 李宏博, 夏盼盼, 刘泽鹏, 夏利宁. 外源MDR鼠伤寒沙门菌对健康小鼠肠道菌群的影响[J]. 畜牧兽医学报, 2023, 54(5): 2158-2169. |
| [10] | 胥辉豪, 刘江渝, 李启卷, 郑小波, 林珈好, 金艺鹏, 林德贵. 己糖激酶2在犬乳腺肿瘤中的表达及预后研究[J]. 畜牧兽医学报, 2023, 54(3): 1310-1324. |
| [11] | 李超, 赵雪艳, 王永军, 王彦平, 任一帆, 李菁璇, 王怀中, 王继英, 宋勤叶. 莱芜猪和杜长大猪盲肠和结肠微生物菌群结构组成和功能分析[J]. 畜牧兽医学报, 2023, 54(12): 5033-5045. |
| [12] | 陈璐, 常新宇, 沈嘉忱, 沈瑞廷, 赵振华, 侯晓林. 鸡传染性支气管炎病毒感染HD11细胞的转录组分析[J]. 畜牧兽医学报, 2023, 54(11): 4860-4865. |
| [13] | 郭銮英, 王妮娜, 李杭远, 纪雨霏, 马骏, 裴明超, 邵建伟, 刘全. 蜱携带牛丙型肝炎病毒新亚型巢式PCR检测方法的建立[J]. 畜牧兽医学报, 2022, 53(3): 972-977. |
| [14] | 任曼, 刘欣, 唐玉林, 张瑞雪, 秦俊杰, 朱浩, 郭延生. 归芪益母复方制剂对产后奶牛瘤胃微生物和短链脂肪酸的调节[J]. 畜牧兽医学报, 2022, 53(12): 4461-4469. |
| [15] | 王乐, 陈泓岑, 张永红, 吴琼, 侯佳佳, 王天祎, 卢天航, 黄传发, 张华, 崔德凤. 基于网络药理学联合16S rDNA高通量测序技术分析丹参对感染大肠杆菌小鼠肠道菌群的影响[J]. 畜牧兽医学报, 2022, 53(10): 3695-3711. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||