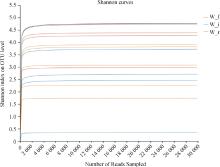
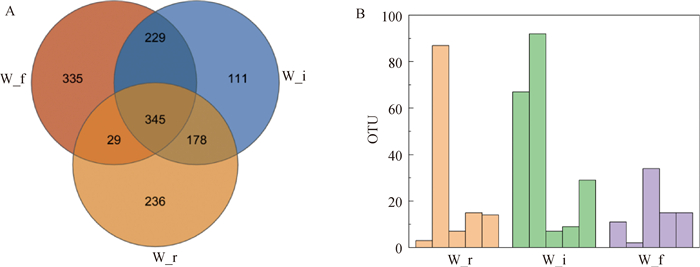
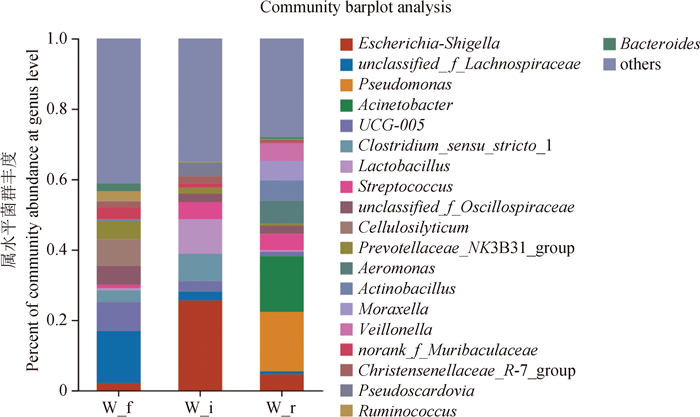
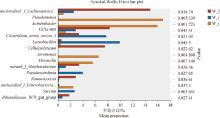
| 1 |
FREDRIKSSON-AHOMAA M . Wild Boar: A reservoir of foodborne zoonoses[J]. Foodborne Pathog Dis, 2019, 16 (3): 153- 165.
doi: 10.1089/fpd.2018.2512
|
| 2 |
周舒枫. 野猪移出名录保护意识不减"三有"野生动物名录调整收录陆生野生动物1924种[J]. 绿化与生活, 2023 (8): 1- 2.
|
|
ZHOU S F . Wild boars are removed from the list and their awareness of protection remains unchanged. The "Three Haves" wild animal list has been adjusted to include 1924 species of terrestrial wild animals[J]. Greening and Life, 2023, 8, 1- 2.
|
| 3 |
SHRESTHA A , UZAL F A , MCCLANE B A . Enterotoxic Clostridia: Clostridium perfringens enteric diseases[J]. Microbiol Spectr, 2018, 6 (5)
doi: 10.1128/microbiolspec.gpp3-0003-2017
|
| 4 |
郑艳敏, 滕臣刚, 韩迪, 等. 一起产气荚膜梭菌引发校园食物中毒的流行病学调查[J]. 中国校医, 2022, 36 (8): 568-570, 602.
|
|
ZHENG Y M , TENG C G , HAN D , et al. Epidemiological investigation on food poisoning caused by Clostridium perfringens in campus[J]. Chinese Journal of School Doctor, 2022, 36 (8): 568-570, 602.
|
| 5 |
王冬经, 吴金措姆, 曾江勇. 牦牛源A型产气荚膜梭菌的分离鉴定及生物学特性研究[J]. 中国畜牧兽医, 2024, 51 (1): 229- 241.
|
|
WANG D J , WU J C M , ZENG J Y . Isolation, identification and biological characteristics analysis of Clostridium perfringens type A from yaks[J]. China Animal Husbandry & Veterinary Medicine, 2024, 51 (1): 229- 241.
|
| 6 |
ZHANG T , ZHANG W T , AI D Y , et al. Prevalence and characterization of Clostridium perfringens in broiler chickens and retail chicken meat in central China[J]. Anaerobe, 2018, 54, 100- 103.
doi: 10.1016/j.anaerobe.2018.08.007
|
| 7 |
MEHDIZADEH GOHARI I , A. NAVARRO M , LI J , et al. Pathogenicity and virulence of Clostridium perfringens[J]. Virulence, 2021, 12 (1): 723- 753.
doi: 10.1080/21505594.2021.1886777
|
| 8 |
KIU R , HALL L J . An update on the human and animal enteric pathogen Clostridium perfringens[J]. Emerg Microbes Infect, 2018, 7 (1): 1- 15.
|
| 9 |
GIBERT M , JOLIVET-REYNAUD C , POPOFF M R . Beta2 toxin, a novel toxin produced by Clostridium perfringens[J]. Gene, 1997, 203 (1): 65- 73.
doi: 10.1016/S0378-1119(97)00493-9
|
| 10 |
VAN ASTEN A J A M , NIKOLAOU G N , GRÖNE A . The occurrence of cpb2-toxigenic Clostridium perfringens and the possible role of the beta2-toxin in enteric disease of domestic animals, wild animals and humans[J]. Vet J, 2010, 183 (2): 135- 140.
doi: 10.1016/j.tvjl.2008.11.005
|
| 11 |
ZHANG Y , HOU Z , MA J . Hemorrhagic enterocolitis and death in two felines (Panthera tigris altaica and Panthera leo) associated with Clostridium perfringens type A[J]. J Zoo Wildl Med, 2012, 43 (2): 394- 396.
doi: 10.1638/2010-0197.1
|
| 12 |
ROOD J I , ADAMS V , LACEY J , et al. Expansion of the Clostridium perfringens toxin-based typing scheme[J]. Anaerobe, 2018, 53, 5- 10.
doi: 10.1016/j.anaerobe.2018.04.011
|
| 13 |
JOST B H , BILLINGTON S J , TRINH H. T , et al. Association of genes encoding beta2 toxin and a collagen binding protein in Clostridium perfringens isolates of porcine origin[J]. Vet Microbiol, 2006, 115 (1-3): 173- 182.
doi: 10.1016/j.vetmic.2006.01.012
|
| 14 |
何仪军, 杨新龙, 明小强. 野猪的生活习性危害及防控对策[J]. 农业技术与装备, 2023 (3): 142- 143.
|
|
HE Y J , YANG X M , MING X Q . Hazards of living habits and prevention and control countermeasures of wild boar[J]. Agricultural Technology & Equipment, 2023 (3): 142- 143.
|
| 15 |
BROWN V R , BOWEN R A , BOSCO-LAUTH A M . Zoonotic pathogens from feral swine that pose a significant threat to public health[J]. Transbound Emerg Dis, 2018, 65 (3): 649- 659.
doi: 10.1111/tbed.12820
|
| 16 |
吴晓傲. 辽宁部分地区野猪病毒性传染病病原谱的构成、主要病原基因特征及qPCR检测方法的建立[D]. 沈阳: 沈阳农业大学, 2023.
|
|
WU X A. Composition of the pathogenic spectrum of viral infectiousdiseases of wild boars in some areas of Liaoning, characterization of the main pathogenic genes andestablishment of qPCR detection methods[D]. Shenyang: Shenyang Agricultural University, 2023. (in Chinese)
|
| 17 |
MENG X J , LINDSAY D S , SRIRANGANATHAN N . Wild boars as sources for infectious diseases in livestock and humans[J]. Philos Trans R Soc Lond B Biol Sci, 2009, 364 (1530): 2697- 2707.
doi: 10.1098/rstb.2009.0086
|
| 18 |
SILVA R O , D'ELIA M L , TEIXEIRA É P T , et al. Clostridium difficile and Clostridium perfringens from wild carnivore species in Brazil[J]. Anaerobe, 2014, 28, 207- 211.
doi: 10.1016/j.anaerobe.2014.06.012
|
| 19 |
韩春生, 戴益民, 王林康, 等. 仔猪粪便DNA中产气荚膜梭菌主要毒素基因检测及荧光定量PCR计数方法的建立[J]. 中国兽医学报, 2019, 39 (1): 57- 62.
|
|
HAN C S , DAI Y M , WANG L K , et al. Detection of major toxin genes and establishment of a real-time PCR assay with quantitative of Clostridium perfringens from the fecal DNA of piglets[J]. Chinese Journal of Veterinary Science, 2019, 39 (1): 87- 62.
|
| 20 |
刘永杰, 王渊, 付强, 等. 高通量测序技术在病原生物学方面的研究进展[J]. 口岸卫生控制, 2019, 24 (1): 6- 9.
|
|
LIU Y J , WANG Y , FU Q , et al. Advances in high-throughput sequencing technology in the field of pathogen biology y[J]. Port Health Control, 2019, 24 (1): 6- 9.
|
| 21 |
SHIN N R , WHON T W , BAE J W . Proteobacteria: microbial signature of dysbiosis in gut microbiota[J]. Trends Biotechnol, 2015, 33 (9): 496- 503.
doi: 10.1016/j.tibtech.2015.06.011
|
| 22 |
姜长津, 陈云, 潘鹏丞, 等. 育肥猪粪便菌群结构及其与生长速度的相关性分析[J]. 中国畜牧兽医, 2022, 49 (3): 924- 931.
|
|
JIANG C J , CHEN Y , PAN P C , et al. Fecal microflora structure and its correlation with growth rate in finishing pigs[J]. China Animal Husbandry & Veterinary Medicine, 2022, 49 (3): 924- 931.
|
| 23 |
SHINKAI T , UEKI T , KOBAYASHI Y . Detection and identification of rumen bacteria constituting a fibrolytic consortium dominated by Fibrobacter succinogenes[J]. Anim Sci J, 2010, 81 (1): 72- 79.
doi: 10.1111/j.1740-0929.2009.00698.x
|
| 24 |
杨雄威, 彭彩淳, 郭群毅, 等. 贵州苗岭地区野猪肠道菌群结构与功能分析[J]. 兽类学报, 2021, 41 (4): 365- 376.
|
|
YANG X W , PENG C C , GUO Q Y , et al. Diversity and functional characteristics of intestinal microflora of free-living wild boars in the Miaoling Mountain area in Guizhou Province, China[J]. Acta Theriologica Sinica, 2021, 41 (4): 365- 376.
|
| 25 |
PIROLO M , ESPINOSA-GONGORA C , ALBERDI A , et al. Bacterial topography of the upper and lower respiratory tract in pigs[J]. Anim Microbiome, 2023, 5 (1): 5.
doi: 10.1186/s42523-023-00226-y
|
| 26 |
陈强. 猪魏氏梭菌病的发病原因、临床症状、剖检变化及防控措施[J]. 现代畜牧科技, 2019 (3): 86- 87.
|
|
CHEN Q . The causes, clinical symptoms, changes in autopsy, and prevention and control measures of porcine Clostridium wilt disease[J]. Modern Animal Husbandry Science and Technology, 2019 (3): 86- 87.
|
| 27 |
LI M , ZHANG X , ZHU L , et al. Identification, isolation, and phylogenetic analysis of Clostridium perfringens Type A and Type C from wild boar (Sus scrofa) in the people's republic of China[J]. J Wildl Dis, 2017, 53 (3): 642- 648.
doi: 10.7589/2016-08-199
|
| 28 |
周姣. 仔猪A型产气荚膜梭菌β2毒素毒性作用及菌株JXJA17全基因组序列分析[D]. 南昌: 江西农业大学, 2018.
|
|
ZHOU J. The analysis of β2 toxin effect harboring by CpA and the whole-genome sequence of strain JXJA17 from piglets[D]. Nanchang: Jiangxi Agricultural University, 2018. (in Chinese)
|
| 29 |
ZENG X , LIU B , ZHOU J , et al. Complete genomic sequence and analysis of β2 toxin gene mapping of Clostridium perfringens JXJA17 isolated from piglets in China[J]. Sci Rep, 2021, 11 (1): 475.
doi: 10.1038/s41598-020-79333-8
|



 ), 杨贝莹1, 武前悦1, 花慧颖1, 曹华斌1, 严珲2, 张锦华1,*(
), 杨贝莹1, 武前悦1, 花慧颖1, 曹华斌1, 严珲2, 张锦华1,*(