


畜牧兽医学报 ›› 2025, Vol. 56 ›› Issue (10): 4947-4962.doi: 10.11843/j.issn.0366-6964.2025.10.016
冯达( ), 魏趁*(), 胡思怡, 杜春梅, 马健, 吴江, 周光现, 甘尚权*()
), 魏趁*(), 胡思怡, 杜春梅, 马健, 吴江, 周光现, 甘尚权*()
收稿日期:2025-03-13
出版日期:2025-10-23
发布日期:2025-11-01
通讯作者:
魏趁,甘尚权
E-mail:fda_1020@163.com;weichenwjf@126.com;shangquangan@gdou.edu.cn
作者简介:冯达(1999-),男,河南濮阳人,硕士生,主要从事羊遗传育种研究,E-mail: fda_1020@163.com
基金资助:
FENG Da(), WEI Chen*(), HU Siyi, DU Chunmei, MA Jian, WU Jiang, ZHOU Guangxian, GAN Shangquan*()
Received:2025-03-13
Online:2025-10-23
Published:2025-11-01
Contact:
WEI Chen, GAN Shangquan
E-mail:fda_1020@163.com;weichenwjf@126.com;shangquangan@gdou.edu.cn
摘要:
旨在解析雷州山羊的群体遗传多样性和遗传结构,为其种质资源的保护与可持续利用提供理论依据。本研究采集了20只雷州山羊的血液样本进行了全基因组重测序,平均测序深度约为10×,同时从NCBI数据库下载了43头山羊的全基因组数据(包括云上黑山羊、金堂黑山羊、济宁青山羊、西藏山羊和隆林山羊)。基于全基因组数据,利用PopLDdecay软件进行了连锁不平衡(LD)分析;利用Plink软件计算了个体间遗传距离(IBS),并通过GCTA软件构建亲缘关系矩阵;此外,采用Plink软件进行主成分分析(PCA),利用Phylip软件构建邻接法(NJ)系统发育树,并通过ADMIXTURE软件进行群体结构分析及可视化,评估山羊群体的群体结构和遗传多样性。最后分析连续性纯合片段(ROH)以及计算群体间遗传分化指数(FST), 对鉴定到的区间进行基因注释和功能富集分析。结果共检测到18 810 921个SNPs,其中大部分位于基因间区(65.71%)。雷州山羊的HO、HE、MAF、Pi、PIC、Ne和FIS分别为0.298、0.295、0.211、0.001 8、0.808、65.488和0.086,其中HO高于HE,表明其群体遗传多样性处于正常水平。LD分析显示,雷州山羊的LD衰减速度最快,表明其群体遗传多样性较高且基因组受选择程度较低。IBS距离矩阵和亲缘关系G矩阵分析结果表明,雷州山羊群体内个体间亲缘关系较近,但与其他山羊群体存在显著遗传分化。ROH结果显示雷州山羊群体具有较低的近交水平。PCA结果显示,雷州山羊群体呈现离散分布且独立于其他群体;NJ系统发育树进一步支持雷州山羊形成独立分支;群体结构分析表明,当K=3时群体结构最佳,此时雷州山羊表现出独特的遗传结构。基于全基因组FST分析,筛选前1%的高分化位点后,发现ADGRL3、CXCR1和HTR1F基因区域存在显著选择信号,提示这些基因可能与适应性相关。本研究全面解析了雷州山羊遗传多样性、亲缘关系和群体遗传结构,为其遗传背景的深入理解提供了科学依据,同时为制定科学的保种策略和开发利用方案奠定了理论基础。
中图分类号:
冯达, 魏趁, 胡思怡, 杜春梅, 马健, 吴江, 周光现, 甘尚权. 基于全基因组重测序分析雷州山羊群体遗传多样性和群体结构[J]. 畜牧兽医学报, 2025, 56(10): 4947-4962.
FENG Da, WEI Chen, HU Siyi, DU Chunmei, MA Jian, WU Jiang, ZHOU Guangxian, GAN Shangquan. Genetic Diversity and Population Structure Analysis of Leizhou Goat Based on Whole Genome Resequencing Analysis[J]. Acta Veterinaria et Zootechnica Sinica, 2025, 56(10): 4947-4962.

图 1
雷州山羊SNPs在染色体上的分布及基因组区域注释情况 A. SNPs在29条染色体上的分布情况,纵坐标为1~29条染色体,横坐标为染色体上每1 Mb的窗口,不同颜色为每1 Mb窗口所含SNPs的数量。B. SNPs的基因组区域注释分布"
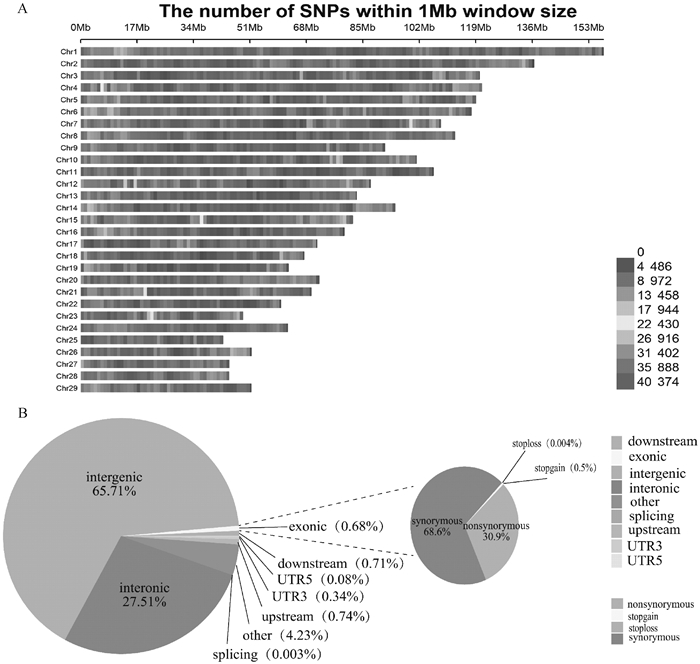
表 1
6个山羊群体的遗传多样性指标"
| 群体 Population | 最小等位基因频率 MAF | 观测杂合度 HO | 期望杂合度 HE | 有效群体含量 Ne | 多态信息含量 PIC | 核苷酸多态性 Pi | 近交系数 FIS |
| 雷州山羊 LZ | 0.211 | 0.298 | 0.295 | 65.488 | 0.808 | 1.798×10-3 | 0.086 |
| 云上黑山羊 YS | 0.207 | 0.305 | 0.290 | 21.312 | 0.938 | 2.179×10-3 | 0.053 |
| 金堂黑山羊 JT | 0.198 | 0.280 | 0.281 | 23.478 | 0.845 | 1.730×10-3 | 0.092 |
| 济宁青山羊 JN | 0.188 | 0.276 | 0.269 | 24.300 | 0.947 | 2.354×10-3 | 0.299 |
| 西藏山羊 TB | 0.188 | 0.250 | 0.270 | 25.879 | 0.947 | 2.080×10-3 | 0.083 |
| 隆林山羊 LL | 0.227 | 0.313 | 0.315 | 16.124 | 0.750 | 2.043×10-3 | 0.075 |

图 2
6个山羊群体的连锁不平衡(LD)衰减 横坐标为物理距离(kb),纵坐标为LD系数(r2)。LZ.雷州山羊;LL.隆林山羊;TB.西藏山羊;JN.济宁青山羊;JT.金堂黑山羊;YS.云上黑山羊,下同"
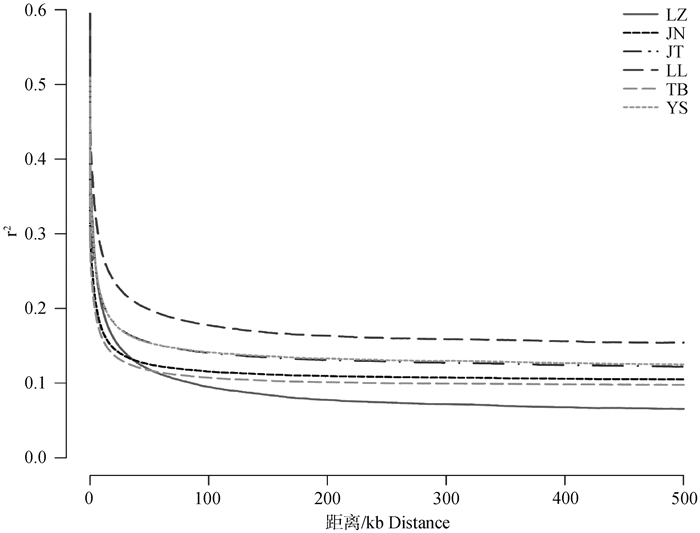

图 3
6个山羊群体的ROH分析 A. 每个个体的ROH数目与ROH长度分布,每个点为一个个体,点的颜色表示个体所属群体,横坐标对应每个个体的ROH数目,纵坐标对应每个个体的ROH长度(Mb)。B. FROH分布的小提琴图,横坐标为各个群体,纵坐标为FROH系数,小提琴宽窄代表个体的分布情况,内部箱线图的箱体部分表示四分位数范围,箱体内部横线表示中位数"
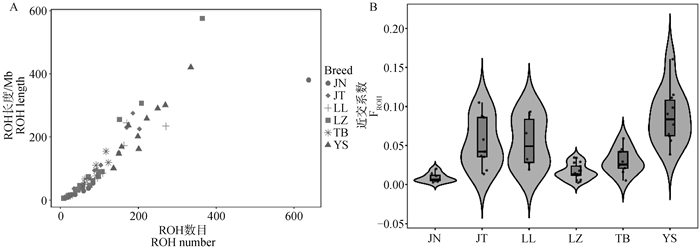

图 4
6个山羊群体的G矩阵分析可视化结果 横、纵坐标为山羊个体,每一个小方格代表两个个体间的亲缘关系,颜色越接近红色表明两个个体间的亲缘关系越近,反之亦然"
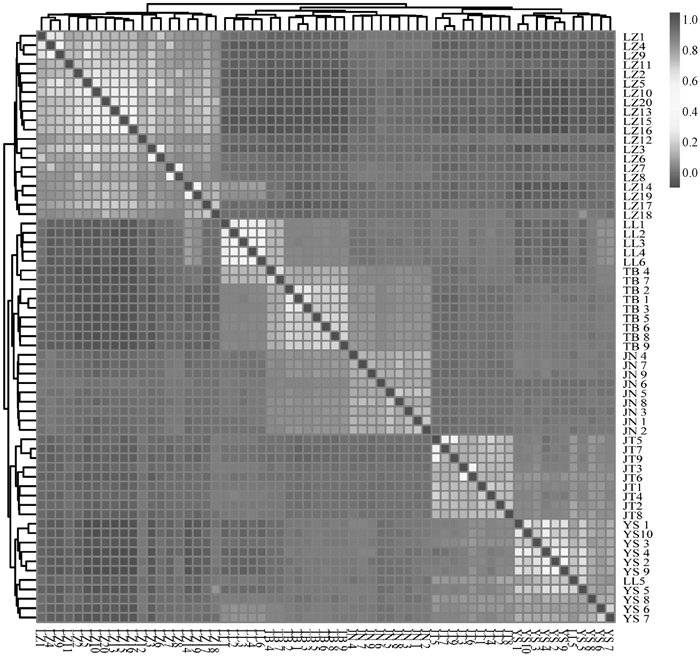

图 5
6个山羊群体的IBS遗传距离矩阵结果 横、纵坐标为山羊个体,每一个小方格代表两个个体间的遗传距离值,颜色越接近红色表明两个个体间的遗传距离越大,反之亦然"
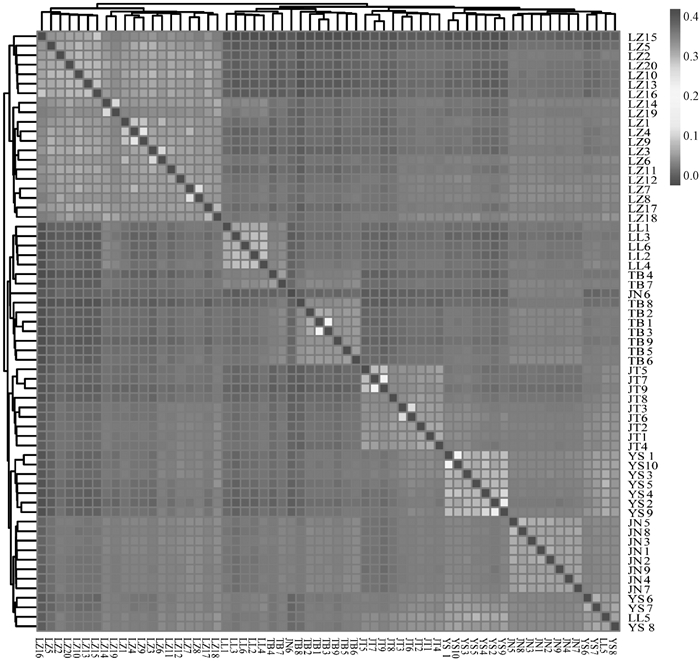
图 6
6个山羊群体的群体结构分析 A. 主成分分析;B. 系统发育树;C. 不同K值的交叉验证错误率;D. Admixture评估K=2、3和6时的群体结构"
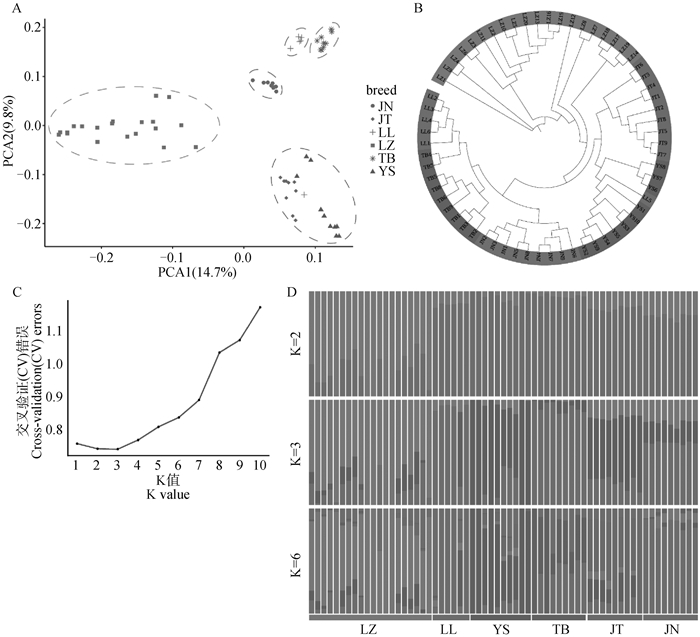
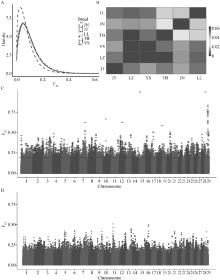
图 7
山羊群体的FST分析 A. FST密度图,x轴为FST指数,不同颜色表示不同群体间的分析。B. FST热图,颜色深浅表示FST指数高低,每个方框表示其对应的x轴群体和y轴群体之间的平均FST值。C. LZ与TB群体FST分析的曼哈顿图。D. LZ与YS群体FST分析的曼哈顿图"
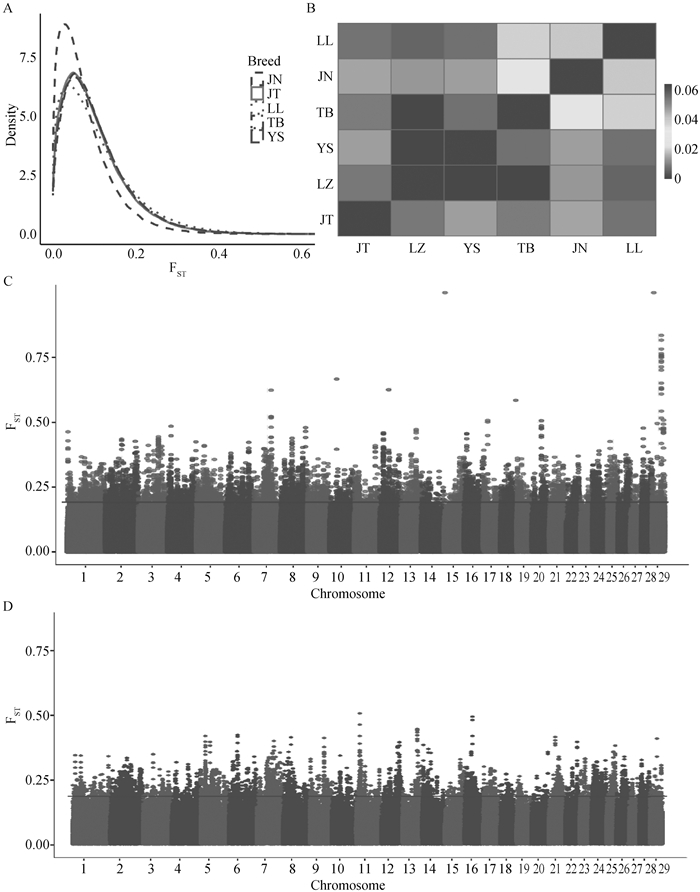
表 2
LZ与TB群体GO功能富集分析"
| GO条目 GO term | P值 Pvalue | 基因 Gene |
| olfactory receptor activity | 1.9×10-4 | LOC102185414, LOC102183215, LOC102183415, LOC102182462, LOC102184860, LOC102183344等 |
| G protein-coupled receptor activity | 2.8×10-4 | ADGRL3, LOC102185414, LOC102183215, LOC102183415, LOC102182462, LOC102184860等 |
| G protein-coupled receptor signaling pathway | 7.1×10-4 | CXCR1, LOC102185414, LOC102183215, LOC102183415, LOC102182462, LOC102184860等 |
| plasma membrane | 0.001 1 | ADGRL3, SHC2, GRIA4, CRIPTO, NPC1, DNAJC5等 |
| odorant binding | 0.002 0 | LOC102185414, LOC102183215, LOC102175834, LOC102169980, LOC102182462, LOC102184860 |
| sensory perception of smell | 0.002 2 | LOC102185414, LOC102183215, LOC102175834, LOC102169980, LOC102182462, LOC102184860 |
| lysozyme activity | 0.003 9 | LOC102185900, LOC102172331, LOC108633237 |
| defense response to Gram-negative bacterium | 0.004 3 | LOC102171106, LOC102185900, LOC102172331, LOC108633237 |
| killing of cells of another organism | 0.009 9 | LOC102185900, LOC102172331, LOC108633237 |

图 8
LZ与TB以及YS群体KEGG通路图 A. LZ与TB群体间FST分析鉴定显著候选基因的KEGG通路图。B. LZ与YS群体间FST分析鉴定显著候选基因的KEGG通路图。纵轴代表KEGG富集通路名,横轴代表富集分数,圆的大小代表富集的基因数量,圆的颜色梯度代表显著性大小"
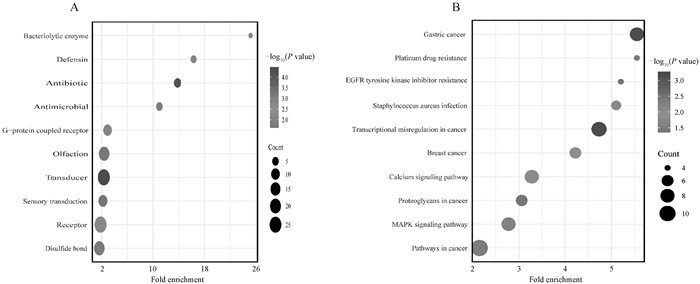
表 3
LZ与YS群体GO功能富集分析"
| GO条目 GO term | P值 P value | 基因 Gene |
| G protein-coupled receptor signaling pathway | 1.1×10-6 | LOC102187858, LOC102182402, LOC102183415, LOC108634193, LOC102186762, LOC102187045等 |
| olfactory receptor activity | 1.9×10-6 | LOC102187858, LOC102182402, LOC102183415, LOC108634193, LOC102186762, LOC102187045等 |
| membrane | 1.2×10-5 | CEBPZOS, UPK3BL2, NUDT4, SEC11C, TMEM254, BST2, NAT8, DNAJC5, OSBPL1A等 |
| G protein-coupled receptor activity | 1.5×10-5 | LOC102187858, LOC102182402, LOC102183415, LOC108634193, LOC102186762, LOC102187045等 |
| structural constituent of eye lens | 0.005 9 | CRYGC, LOC102191000, LOC102188265 |
| lens development in camera-type eye | 0.011 | CRYGC, LOC102191000, LOC102188265 |
| cyanamide hydratase activity | 0.019 | LOC106502860, LOC108637506 |
| plasma membrane | 0.028 | HTR1F, SHC2, PIEZO2, GRIA4, SYTL2, DNAJC5, OSBPL1A |
| Glycerophosphodiester phosphodiesterase activity | 0.031 | LOC102183767, LOC102183296 |
| lipid transport | 0.047 | OSBPL1A, LOC102172041, LOC102172606 |
表 4
适应性相关候选基因"
| 染色体 Chromosome | 基因 Gene | 编号 ID | 描述 Description |
| 2 | ADGRL3 | ENSCHIG00000012101 | Adhesion G Protein-Coupled Receptor L3 |
| 6 | CXCR1 | ENSCHIG00000020632 | C-X-C motif chemokine receptor 1 |
| 1 | HTR1F | ENSCHIG00000003029 | 5-hydroxytryptamine receptor 1F |
| 1 | 国家畜禽遗传资源委员会.中国畜禽遗传资源志(羊志)[M].北京:中国农业出版社,2011. |
| China National Commission of Animal Genetic Resources.Animal genetic resources in China(Sheep and Goats)[M].Beijing:China Agriculture Press,2011. | |
| 2 |
KICHAMUN,WANJALAG,CZISZTERL T,et al.Assessing the population structure and genetic variability of kenyan native goats under extensive production system[J].Sci Rep,2024,14(1):16342.
doi: 10.1038/s41598-024-67374-2 |
| 3 |
MUKHINAV,SVISHCHEVAG,VORONKOVAV,et al.Genetic diversity, population structure and phylogeny of indigenous goats of mongolia revealed by SNP genotyping[J].Animals,2022,12(3):221.
doi: 10.3390/ani12030221 |
| 4 |
VISSERC,LASHMARS F,VAN MARLE-KOSTERE,et al.Genetic diversity and population structure in south african, french and argentinian angora goats from genome-wide SNP data[J].PLoS ONE,2016,11(5):e0154353.
doi: 10.1371/journal.pone.0154353 |
| 5 |
VAN DIJKE L,AUGERH,JASZCZYSZYNY,et al.Ten years of next-generation sequencing technology[J].Trends Genet,2014,30(9):418-426.
doi: 10.1016/j.tig.2014.07.001 |
| 6 | GOODWINS,MCPHERSONJ D,MCCOMBIEW R.Coming of age: Ten years of next-generation sequencing technologies[J].Nat Rev Genet,2016,17(6):333-351. |
| 7 | SHASTRYB S.SNPs: Impact on Gene Function and Phenotype[J].Methods Mol Biol,2009,578,3-22. |
| 8 |
ZHUZ,ZHANGL,XINQ,et al.Whole-genome sequencing revealed the population structure of fujian chicken breeds[J].Czech J Anim Sci,2024,69(8):323-330.
doi: 10.17221/91/2023-CJAS |
| 9 |
TONGX,HOUL,HEW,et al.Whole genome sequence analysis reveals genetic structure and X-chromosome haplotype structure in indigenous Chinese pigs[J].Sci Rep,2020,10(1):9433.
doi: 10.1038/s41598-020-66061-2 |
| 10 |
ZHANGY,WEIZ,ZHANGM,et al.Population structure and selection signal analysis of nanyang cattle based on whole-genome sequencing data[J].Genes,2024,15(3):351.
doi: 10.3390/genes15030351 |
| 11 |
SHIH,LIT,SUM,et al.Whole genome sequencing revealed genetic diversity, population structure, and selective signature of panou tibetan sheep[J].BMC Genomics,2023,24(1):50.
doi: 10.1186/s12864-023-09146-2 |
| 12 |
CHANGL,ZHENGY,LIS,et al.Identification of genomic characteristics and selective signals in guizhou black goat[J].BMC Genomics,2024,25(1):164.
doi: 10.1186/s12864-023-09954-6 |
| 13 |
CHENQ,HUANGY,WANGZ,et al.Whole-genome resequencing reveals diversity and selective signals in Longlin goat[J].Gene,2021,771,145371.
doi: 10.1016/j.gene.2020.145371 |
| 14 |
EG X,ZHAOY J,CHENL P,et al.Genetic diversity of the Chinese goat in the littoral zone of the yangtze river as assessed by microsatellite and mtDNA[J].Ecol Evol,2018,8(10):5111-5123.
doi: 10.1002/ece3.4100 |
| 15 | 郭家中,张一菲,武国,等.利用全基因组重测序分析雅安奶山羊遗传多样性和遗传结构[J].四川农业大学学报,2025,43(2):444-451. |
| GUOJ Z,ZHANGY F,WUG,et al.Analysis on genetic diversity and genetic structure of Yaan dairy goats based on whole-genome resequencing[J].Journal of Sichuan Agricultural University,2025,43(2):444-451. | |
| 16 |
MAK,LID,QIX,et al.Population structure, runs of homozygosity analysis and construction of single nucleotide polymorphism fingerprinting database of Longnan goat population[J].Food Energy Secur,2024,13(1):e517.
doi: 10.1002/fes3.517 |
| 17 |
SHERIFFO,AHBARAA M,HAILEA,et al.Whole-genome resequencing reveals genomic variation and dynamics in Ethiopian indigenous goats[J].Front Genet,2024,15,1353026.
doi: 10.3389/fgene.2024.1353026 |
| 18 |
CHENS,ZHOUY,CHENY,et al.fastp: An ultra-fast all-in-one FASTQ preprocessor[J].Bioinformatics,2018,34(17):i884-i890.
doi: 10.1093/bioinformatics/bty560 |
| 19 |
LIH,DURBINR.Fast and accurate short read alignment with burrows-wheeler transform[J].Bioinformatics,2009,25(14):1754-1760.
doi: 10.1093/bioinformatics/btp324 |
| 20 |
BICKHARTD M,ROSENB D,KORENS,et al.Single-molecule sequencing and chromatin conformation capture enable de novo reference assembly of the domestic goat genome[J].Nat Genet,2017,49(4):643-650.
doi: 10.1038/ng.3802 |
| 21 |
LIH,HANDSAKERB,WYSOKERA,et al.The sequence alignment/map format and SAMtools[J].Bioinformatics,2009,25(16):2078-2079.
doi: 10.1093/bioinformatics/btp352 |
| 22 |
MCKENNAA,HANNAM,BANKSE,et al.The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data[J].Genome Res,2010,20(9):1297-1303.
doi: 10.1101/gr.107524.110 |
| 23 |
PEDERSENB S,QUINLANA R.Mosdepth: Quick coverage calculation for genomes and exomes[J].Bioinformatics,2018,34(5):867-868.
doi: 10.1093/bioinformatics/btx699 |
| 24 |
YANGH,WANGK.Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR[J].Nat Protoc,2015,10(10):1556-1566.
doi: 10.1038/nprot.2015.105 |
| 25 |
PURCELLS,NEALEB,TODD-BROWNK,et al.PLINK: A tool set for whole-genome association and population-based linkage analyses[J].Am J Hum Genet,2007,81(3):559-575.
doi: 10.1086/519795 |
| 26 |
DANECEKP,AUTONA,ABECASISG,et al.The variant call format and VCFtools[J].Bioinformatics,2011,27(15):2156-2158.
doi: 10.1093/bioinformatics/btr330 |
| 27 |
ZHANGC,DONGS S,XUJ Y,et al.PopLDdecay: A fast and effective tool for linkage disequilibrium decay analysis based on variant call format files[J].Bioinformatics,2019,35(10):1786-1788.
doi: 10.1093/bioinformatics/bty875 |
| 28 |
YANGJ,LEES H,GODDARDM E,et al.GCTA: A tool for genome-wide complex trait analysis[J].Am J Hum Genet,2011,88(1):76-82.
doi: 10.1016/j.ajhg.2010.11.011 |
| 29 |
YANGJ,BENYAMINB,MCEVOYB P,et al.Common SNPs explain a large proportion of the heritability for human height[J].Nat Genet,2010,42(7):565-569.
doi: 10.1038/ng.608 |
| 30 | VILLANUEVAR A M,CHENZ J.ggplot2: Elegant graphics for data analysis (2nd ed.)[J].Meas-Interdiscip Res,2019,17(3):160-167. |
| 31 |
LEET H,GUOH,WANGX,et al.SNPhylo: a pipeline to construct a phylogenetic tree from huge SNP data[J].BMC Genomics,2014,15(1):162.
doi: 10.1186/1471-2164-15-162 |
| 32 |
ALEXANDERD H,NOVEMBREJ,LANGEK.Fast model-based estimation of ancestry in unrelated individuals[J].Genome Res,2009,19(9):1655-1664.
doi: 10.1101/gr.094052.109 |
| 33 |
QUINLANA R,HALLI M.BEDTools: A flexible suite of utilities for comparing genomic features[J].Bioinformatics,2010,26(6):841-842.
doi: 10.1093/bioinformatics/btq033 |
| 34 |
SHERMANB T,HAOM,QIUJ,et al.DAVID: a web server for functional enrichment analysis and functional annotation of gene lists (2021 update)[J].Nucleic Acids Res,2022,50(W1):W216-W221.
doi: 10.1093/nar/gkac194 |
| 35 |
COLLIL,MILANESIM,TALENTIA,et al.Genome-wide SNP profiling of worldwide goat populations reveals strong partitioning of diversity and highlights post-domestication migration routes[J].Genet Sel Evol,2018,50(1):58.
doi: 10.1186/s12711-018-0422-x |
| 36 |
SUNX,GUOJ,LIL,et al.Genetic diversity and selection signatures in jianchang black goats revealed by whole-genome sequencing data[J].Animals,2022,12(18):2365.
doi: 10.3390/ani12182365 |
| 37 |
ZHANGT,WANGZ,LIY,et al.Genetic diversity and population structure in five inner mongolia cashmere goat populations using whole-genome genotyping[J].Anim Biosci,2024,37(7):1168-1176.
doi: 10.5713/ab.23.0424 |
| 38 |
GEBRESELASEH B,NIGUSSIEH,WANGC,et al.Genetic diversity, population structure and selection signature in begait goats revealed by whole-genome sequencing[J].Animals,2024,14(2):307.
doi: 10.3390/ani14020307 |
| 39 |
WEIC,LUJ,XUL,et al.Genetic structure of Chinese indigenous goats and the special geographical structure in the southwest China as a geographic barrier driving the fragmentation of a large population[J].PLoS ONE,2014,9(4):e94435.
doi: 10.1371/journal.pone.0094435 |
| 40 |
CAIY,FUW,CAID,et al.Ancient genomes reveal the evolutionary history and origin of cashmere-producing goats in China[J].Mol Biol Evol,2020,37(7):2099-2109.
doi: 10.1093/molbev/msaa103 |
| 41 |
CEBALLOSF C,JOSHIP K,CLARKD W,et al.Runs of homozygosity: Windows into population history and trait architecture[J].Nat Rev Genet,2018,19(4):220-234.
doi: 10.1038/nrg.2017.109 |
| 42 |
KELLERM C,VISSCHERP M,GODDARDM E.Quantification of inbreeding due to distant ancestors and its detection using dense single nucleotide polymorphism data[J].Genetics,2011,189(1):237-249.
doi: 10.1534/genetics.111.130922 |
| 43 |
HUANGC,ZHAOQ,CHENQ,et al.Runs of homozygosity detection and selection signature analysis for local goat breeds in yunnan, China[J].Genes,2024,15(3):313.
doi: 10.3390/genes15030313 |
| 44 |
SHIL,WANGL,LIUJ,et al.Estimation of inbreeding and identification of regions under heavy selection based on runs of homozygosity in a large white pig population[J].J Anim Sci Biotechnol,2020,11(1):46.
doi: 10.1186/s40104-020-00447-0 |
| 45 |
FREITASP H F,WANGY,YANP,et al.Genetic diversity and signatures of selection for thermal stress in cattle and other two bos species adapted to divergent climatic conditions[J].Front Genet,2021,12,604823.
doi: 10.3389/fgene.2021.604823 |
| 46 |
WANGJ.Marker-based estimates of relatedness and inbreeding coefficients: an assessment of current methods[J].J Evol Biol,2014,27(3):518-530.
doi: 10.1111/jeb.12315 |
| 47 |
LIG,TANGJ,HUANGJ,et al.Genome-wide estimates of runs of homozygosity, heterozygosity, and genetic load in two chinese indigenous goat breeds[J].Front Genet,2022,13,774196.
doi: 10.3389/fgene.2022.774196 |
| 48 |
王浩宇,马克岩,李讨讨,等.基于简化基因组测序评估小骨山羊群体遗传多样性和群体结构[J].畜牧兽医学报,2025,56(3):1170-1179.
doi: 10.11843/j.issn.0366-6964.2025.03.018 |
|
WANGH Y,MAK Y,LIT T,et al.Population genetic diversity and population structure analysis of Small-boned goat based on specific-locus amplified fragment sequencing[J].Acta Veterinaria et Zootechnica Sinica,2025,56(3):1170-1179.
doi: 10.11843/j.issn.0366-6964.2025.03.018 |
|
| 49 | JOLLIFFEI T,CADIMAJ.Principal component analysis: a review and recent developments[J].Philo Trans A Math Phys Eng Sci,2016,374(2065):20150202. |
| 50 |
KAPLIP,YANGZ,TELFORDM J.Phylogenetic tree building in the genomic age[J].Nat Rev Genet,2020,21(7):428-444.
doi: 10.1038/s41576-020-0233-0 |
| 51 |
WANGS,DELEONC,SUNW,et al.Alternative splicing of latrophilin-3 controls synapse formation[J].Nature,2024,626(7997):128-135.
doi: 10.1038/s41586-023-06913-9 |
| 52 |
TOYAS,STRUYFS,HUERTAL,et al.A narrative review of chemokine receptors CXCR1 and CXCR2 and their role in acute respiratory distress syndrome[J].Eur Respir Rev,2024,33(173):230172.
doi: 10.1183/16000617.0172-2023 |
| 53 |
HUANGS,XUP,SHEND D,et al.GPCRs steer gi and gs selectivity via TM5-TM6 switches as revealed by structures of serotonin receptors[J].Mol Cell,2022,82(14):2681-2695.e6.
doi: 10.1016/j.molcel.2022.05.031 |
| 54 |
WENX,SHANGP,CHENH,et al.Evolutionary study and structural basis of proton sensing by mus GPR4 and xenopus GPR4[J].Cell,2025,188(3):653-670.e24.
doi: 10.1016/j.cell.2024.12.001 |
| [1] | 刘莎, 杨彩春, 张晓雨, 陈琼, 刘雄, 陈洪波, 周焕焕, 史良玉. 基于80K SNP芯片的梅花星猪群体遗传结构解析及全基因组连续纯合片段特征研究[J]. 畜牧兽医学报, 2025, 56(8): 3749-3760. |
| [2] | 任千姿, 张佰忠, 王真勍, 王向林, 龚颖, 胡仁科, 浦亚斌, 苏鹏, 李业芳, 马月辉, 李昊帮, 蒋琳. 基于全基因组重测序对武雪山羊的遗传进化分析[J]. 畜牧兽医学报, 2025, 56(8): 3787-3801. |
| [3] | 缪俊杰, 张日泉, 吴厚义, 游新明, 黄奕雯, 黄小英, 郭震洋, 刘建林, 肖卫华, 郭田华, 陈浩, 康冬柳. 全基因组SNPs揭示井冈黑掌鹅种质资源特性与遗传多样性特征[J]. 畜牧兽医学报, 2025, 56(7): 3199-3209. |
| [4] | 王勤倩, 高振东, 陆颖, 马若珊, 邓卫东, 和晓明. 全基因组重测序在中国地方黄牛上的研究进展[J]. 畜牧兽医学报, 2025, 56(5): 2026-2037. |
| [5] | 姚婷婷, 李昊, 阎卉萱, 曹一凡, 次仁罗布, 索朗曲吉, 尼玛仓决, 赵丽, 旦增洛桑, 斯朗旺姆, 巴桑珠扎, 陈宁博. 西藏自治区10个黄牛群体的mtDNA遗传多样性与母系起源研究[J]. 畜牧兽医学报, 2025, 56(5): 2194-2202. |
| [6] | 王浩宇, 马克岩, 李讨讨, 栗登攀, 赵箐, 马友记. 基于简化基因组测序评估小骨山羊群体遗传多样性和群体结构[J]. 畜牧兽医学报, 2025, 56(3): 1170-1179. |
| [7] | 胡鑫, 游伟, 姜富贵, 成海建, 孙志刚, 宋恩亮. 基于全基因组重测序分析西门塔尔牛遗传多样性与群体结构[J]. 畜牧兽医学报, 2025, 56(3): 1189-1202. |
| [8] | 李亚旋, 邵长亮, 高浩冉, 伍金山, 徐梦琦, 王一鹏, 刘皓君, 苏靖宇, 陈俊华, 李梦欣, 马英杰, 单文娟. 卡拉麦里山蒙古野驴遗传多样性与遗传结构分析[J]. 畜牧兽医学报, 2025, 56(10): 4973-4987. |
| [9] | 刘思宇, 张曼, 张岩, 魏稚彤, 祁兴磊, 高腾云, 刘贤, 梁栋, 付彤. 基于重测序数据评估南阳牛保种效果[J]. 畜牧兽医学报, 2024, 55(9): 3876-3886. |
| [10] | 张涛, 李佳芪, 胥磊, 王丹, 张梦华, 张涛, 闫梦婕, 王玮韬, 范守民, 黄锡霞. 基于全基因组重测序数据的新疆褐牛基因组结构变异检测及群体结构分析[J]. 畜牧兽医学报, 2024, 55(8): 3427-3435. |
| [11] | 宋科林, 闫尊强, 王鹏飞, 程文昊, 李杰, 白雅琴, 孙国虎, 滚双宝. 基于SNP芯片分析徽县青泥黑猪遗传多样性和遗传结构[J]. 畜牧兽医学报, 2024, 55(3): 995-1006. |
| [12] | 任钰为, 陈星, 林燕宁, 黄潇仙, 洪玲玲, 王峰, 孙瑞萍, 张艳, 刘海隆, 郑心力, 晁哲. 基于全基因组重测序研究文昌鸡产蛋性能的影响因素[J]. 畜牧兽医学报, 2024, 55(2): 502-514. |
| [13] | 程昕琰, 王诗媛, 吉叶标, 黄思秀, 杨杰, 孟繁明, 张茂, 蔡更元, 刘琅青. 基于50K SNP芯片评估广东省四类地方猪保种群体的遗传结构[J]. 畜牧兽医学报, 2024, 55(12): 5464-5477. |
| [14] | 徐扩卫, 李卓辉, 冷堂健, 熊宝, 周杰珑, 郭盘江, 王禹, 陈粉粉. 基于全基因组重测序SNP分析宁蒗高原鸡保种群的群体遗传多样性和群体遗传结构[J]. 畜牧兽医学报, 2024, 55(12): 5498-5510. |
| [15] | 梁慧丽, 解玉静, 司博文, 王桂英, 姜运良, 曹贵玲. 基于全基因组重测序分析大尾寒羊基因组变异特征和群体结构[J]. 畜牧兽医学报, 2024, 55(11): 4968-4979. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||