


畜牧兽医学报 ›› 2024, Vol. 55 ›› Issue (12): 5549-5566.doi: 10.11843/j.issn.0366-6964.2024.12.020
闵祥玉( ), 卫佳丽, 许彪, 刘汇涛, 郑军军*(), 王桂武*()
), 卫佳丽, 许彪, 刘汇涛, 郑军军*(), 王桂武*()
收稿日期:2024-03-14
出版日期:2024-12-23
发布日期:2024-12-27
通讯作者:
郑军军,王桂武
E-mail:mxy10504@163.com;zhengjunjun@caas.cn;wangguiwu2005@163.com
作者简介:闵祥玉(1998-), 女, 山东潍坊人, 硕士生, 主要从事动物繁育原理与技术研究, E-mail: mxy10504@163.com
基金资助:
MIN Xiangyu(), WEI Jiali, XU Biao, LIU Huitao, ZHENG Junjun*(), WANG Guiwu*()
Received:2024-03-14
Online:2024-12-23
Published:2024-12-27
Contact:
ZHENG Junjun, WANG Guiwu
E-mail:mxy10504@163.com;zhengjunjun@caas.cn;wangguiwu2005@163.com
摘要:
旨在获得梅花鹿鹿茸组织全长转录本和结构信息,并进一步挖掘出影响梅花鹿鹿茸产量的功能基因。本研究采取饲养管理条件一致的6头2锯龄健康雄性东大梅花鹿,取其相同部位鹿茸组织。根据茸重将鹿茸分为高、低产两组,每组3个重复。利用PacBio Sequel平台以及Illumina平台分别对高、低产鹿茸混合样品进行全长转录组测序。结合高、低产两组二代测序技术,使用SUPPA、KOBOS-I、Cytoscape等工具对可变剪接、功能富集及Hub基因等预测分析。高、低产组测序分别获得有效插入片段36 074 385 bp和32 413 962 bp;转录因子注释显示高产组中bHLH家族转录本数量要高于TF_bZIP家族,低产组中则相反。高产鹿茸组共存在6 644个可变剪接事件,而低产鹿茸组则为7 626个可变剪接事件;经比对,COL1A1在多个融合基因中起主效作用;两组样品中共筛选出207个差异表达基因,KEGG富集显示差异表达基因主要集中在甲状旁腺激素的合成、分泌以及胆固醇代谢等通路;鉴定到11个Hub基因,分别是APOB、SH3GL2、SYT1、ANGPTL4、FGF21、CACNA1E、SERPINF2、CACNB4、KLK4、LRP2、KCNA1。本研究结果为进一步的功能基因鉴定工作和分子育种策略的发展提供了新的理论基础和研究路径。
中图分类号:
闵祥玉, 卫佳丽, 许彪, 刘汇涛, 郑军军, 王桂武. 梅花鹿鹿茸全长转录组测序及鹿茸产量相关基因的挖掘[J]. 畜牧兽医学报, 2024, 55(12): 5549-5566.
MIN Xiangyu, WEI Jiali, XU Biao, LIU Huitao, ZHENG Junjun, WANG Guiwu. Full-length Transcriptome Sequencing of Sika Deer Antler and Mining of Antler Yield-related Genes[J]. Acta Veterinaria et Zootechnica Sinica, 2024, 55(12): 5549-5566.
表 1
梅花鹿二杠茸样品重量信息表"
| 样品名称Sample name | H1 | H2 | H3 | L1 | L2 | L3 |
| 单支重量Single weight | 670 | 777 | 780 | 234 | 216 | 291 |

图 1
高、低产组梅花鹿茸重差异分析 ***. P < 0.01"

表 2
q-PCR引物序列表"
| 引物名称 Primer name | 序列(5′→3′) Sequence | 产物大小/bp Product length | 退火温度/℃ Tm |
| APOB | F: GACTCCAGCCCTCAACTTCC | 183 | 60 |
| R: GTGTTGCCGCCAGTATAGGA | |||
| CACNB4 | F: CACAGCAACCACTCCACAGA | 114 | 60 |
| R: CTGGAAGACAAGCGGTTCCT | |||
| FGF21 | F: CAGCCATCTGAGAACCTGCT | 105 | 60 |
| R: CTGGGGTCTGGGGTTTGAAG | |||
| ANGPTL4 | F: CGGCCCAGCTCACAATATCA | 134 | 60 |
| R: TTACAGTCCAGCCTCCGTCT | |||
| SERPINF2 | F: ATGACACAGTGTTGCTTCTCCT | 151 | 60 |
| R: AGTGGGTATGTGAGGGCTTG | |||
| SYT1 | F: CTACGCCACCCATCACAAGT | 102 | 60 |
| R: CAGAAGGGTACGTGTGGCTT | |||
| GAPDH | F: ATGTTTGTGATGGGCGTGAAC | 265 | 60 |
| R: CCAGTAGAAGCAGGGATGATGTT |

图 2
NR库前20物种分布图"


图 3
KOG功能分类图"


图 4
KEGG功能分类图"


图 5
GO功能分类图 A.高产组GO功能分类图; B. 低产组GO功能分类图"

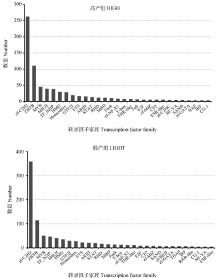
图 6
转录因子家族分布图"

表 3
不同可变剪接事件频数统计"
| 样品名称 Sample | 注释到基因总数 Total genes | 外显子跳跃 SE | 外显子互斥 MX | 内含子滞留 RI | 5′端可变剪接 A5 | 3′端可变剪接 A3 | 起始外显子可变剪接 AF | 终止外显子可变剪接 AL |
| HIGH | 6 644 | 1 361 | 123 | 728 | 753 | 901 | 1 005 | 280 |
| LIGHT | 7 626 | 1 561 | 124 | 1 051 | 952 | 1 026 | 1 164 | 324 |

图 7
差异表达基因分析火山图"
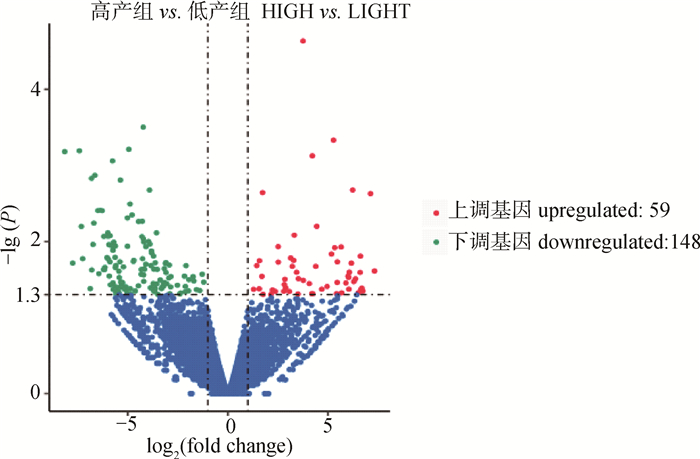

图 8
差异基因KEGG功能富集(P≤0.05)"


图 9
差异基因GO功能富集(P≤0.05)"


图 10
PPI蛋白互作网络 内圈为11个Hub基因"


图 11
qRT-PCR验证"

| 1 |
LIC Y,YANGF H,SHEPPARDA.Adult stem cells and mammalian epimorphic regeneration-insights from studying annual renewal of deer antlers[J].Curr Stem Cell Res Ther,2009,4(3):237-251.
doi: 10.2174/157488809789057446 |
| 2 |
LIC Y,LIY,WANGW Y,et al.Deer antlers: the fastest growing tissue with least cancer occurrence[J].Cell Death Differ,2023,30(12):2452-2461.
doi: 10.1038/s41418-023-01231-z |
| 3 |
ZHANGP,GUOZ H,MAL,et al.Investigation of anti-fatigue effect and simultaneous determination of eight nucleosides in different parts of velvet antler in red deer and sika deer[J].Chem Biodivers,2020,17(2):e1900512.
doi: 10.1002/cbdv.201900512 |
| 4 | 胡艳红,颜鑫,雷燕,等.鹿茸的化学成分、药理作用与临床应用研究进展[J].辽宁中医药大学学报,2021,23(9):47-52. |
| HUY H,YANX,LEIY,et al.Research progress on chemical constituents, pharmacological effects and clinical application of Lurong (Cervi Cornu Pantotrichum)[J].Journal of Liaoning University of TCM,2021,23(9):47-52. | |
| 5 | 李光玉. 鹿茸生长与血液IGF-1浓度年周期变化规律[D]. 长春: 中国农业科学院特产研究所, 2003. |
| LI G Y. Changing law of annual cycle of antler growth and blood IGF-1 concentration[D]. Changchun: Institute of Special Animal and Plant Sciences of CAAS, 2003. (in Chinese) | |
| 6 |
张芙蕊,韩若冰,郭梦雅,等.梅花鹿茸生长过程中顶端不同组织COL1A1基因启动子DNA甲基化模式及差异分析[J].中国农业大学学报,2022,27(10):153-163.
doi: 10.11841/j.issn.1007-4333.2022.10.14 |
|
ZHANGF R,HANR B,GUOM Y,et al.DNA methylation and differential analysis of COL1A1 gene promoter in the different tissues of antler top during the antler growing in sika deer (Cervus nippon)[J].Journal of China Agricultural University,2022,27(10):153-163.
doi: 10.11841/j.issn.1007-4333.2022.10.14 |
|
| 7 | 郭梦雅,韩若冰,邢海华,等.梅花鹿KGF的基因克隆及在不同时期顶端茸皮组织的表达分析[J].中国农业大学学报,2022,27(6):154-161. |
| GUOM Y,HANR B,XINGH H,et al.Cloning of KGF gene from sika deer and expression analysis of antler tip skin tissues at different stages[J].Journal of China Agricultural University,2022,27(6):154-161. | |
| 8 |
BAH X,WANGX,WANGD T,et al.Single-cell transcriptome reveals core cell populations and androgen-RXFP2 axis involved in deer antler full regeneration[J].Cell Regen,2022,11(1):43.
doi: 10.1186/s13619-022-00153-4 |
| 9 | 熊丽容. 梅花鹿MTNR1A基因多态性及其与产茸量的相关性研究[D]. 武汉: 华中农业大学, 2011. |
| XIONG L R. The study of MTNR1A gene in the influence of sika deer and its polymorphism[D]. Wuhan: Huazhong Agricultural University, 2011. (in Chinese) | |
| 10 | 刘慧,王桂武,王权威,等.梅花鹿YWHAE基因编码区全长cDNA的克隆及序列分析[J].农业生物技术学报,2017,25(5):833-841. |
| LIUH,WANGG W,WANGQ W,et al.cDNA coding region cloning and sequence analysing of sika deer (Cervus nippon) YWHAE Gene[J].Journal of Agricultural Biotechnology,2017,25(5):833-841. | |
| 11 |
PARKH J,LEED H,PARKS G,et al.Proteome analysis of red deer antlers[J].Proteomics,2004,4(11):3642-3653.
doi: 10.1002/pmic.200401027 |
| 12 | 鞠妍,刘华淼,何金明,等.基于高通量测序技术的敖鲁古雅驯鹿鹿茸转录组分析[J].中国畜牧兽医,2020,47(7):1981-1989. |
| JUY,LIUH M,HEJ M,et al.Transcriptome analysis of antler in Aoluguya reindeer based on high-throughput sequencing technology[J].China Animal Husbandry & Veterinary Medicine,2020,47(7):1981-1989. | |
| 13 | 杨晓光. 马鹿(Cervus elaphus)鹿茸角顶端茸皮与软骨组织转录组研究[D]. 哈尔滨: 东北林业大学, 2015. |
| YANG X G. Velvet skin and cartilage transcriptomes at the antler tips of Red deer Cervus elaphus[D]. Harbin: Northeast Forestry University, 2015. (in Chinese) | |
| 14 | 幺宝金. 梅花鹿鹿茸顶端组织转录组分析及不同生长期差异基因筛选[D]. 长春: 吉林大学, 2012. |
| YAO B J. Transcriptome analysis and differentially expressed genes screening of antler tips at different growth stages[D]. Changchun: Jilin University, 2012. (in Chinese) | |
| 15 | 崔迎迎,芒来,李蓓,等.MEI1基因可变剪切事件对蒙古马精子生成的调控作用[J].畜牧兽医学报,2022,53(4):1096-1108. |
| CUIY Y,MANGL,LIB,et al.Regulatory role of MEI1 with alternative splicing event on spermatogenesis in mongolian horse[J].Acta Veterinaria et Zootechnica Sinica,2022,53(4):1096-1108. | |
| 16 | 罗朋娜. 鸡GLUT4基因可变剪切体鉴定与主要转录本的功能分析[D]. 郑州: 河南农业大学, 2023. |
| LUO P N. Identification of alternative splicing variants and functional analysis of major transcripts of GUT4 gene in chicken[D]. Zhengzhou: Henan Agricultural University, 2023. (in Chinese) | |
| 17 | 王进娥,麻俊渊,陈舟,等.牦牛发情期与妊娠期转录本及剪切事件研究[J].甘肃农业大学学报,2022,57(6):1-9. |
| WANGJ E,MAJ Y,CHENZ,et al.Transcription and splicing events during estrus and pregnancy in the yak[J].Journal of Gansu Agricultural University,2022,57(6):1-9. | |
| 18 |
陈媛,蔡禾,李利,等.山羊TNNT3基因可变剪切及其对骨骼肌细胞分化的作用[J].中国农业科学,2021,54(20):4466-4477.
doi: 10.3864/j.issn.0578-1752.2021.20.019 |
|
CHENY,CAIH,LIL,et al.Alternative splicing of TNNT3 and its effect on the differentiation of Mu SCs in goat[J].Scientia Agricultura Sinica,2021,54(20):4466-4477.
doi: 10.3864/j.issn.0578-1752.2021.20.019 |
|
| 19 |
HESTANDM S,AMEURA.The versatility of SMRT sequencing[J].Genes (Basel),2019,10(1):24.
doi: 10.3390/genes10010024 |
| 20 |
GASPERINIM,HILLA J,MCFALINE-FIGUEROAJ L,et al.A genome-wide framework for mapping gene regulation via cellular genetic screens[J].Cell,2019,176(1-2):377-390.
doi: 10.1016/j.cell.2018.11.029 |
| 21 |
XINGX M,AIC,WANGT J,et al.The first high-quality reference genome of sika deer provides insights into high-tannin adaptation[J].Genom Proteom Bioinform,2023,21(1):203-215.
doi: 10.1016/j.gpb.2022.05.008 |
| 22 | ALAMANCOSG P,PAGōSA,TRINCADOJ L,et al.Leveraging transcript quantification for fast computation of alternative splicing profiles[J].Proc Natl Acad Sci U S A,2015,21(9):1521-1531. |
| 23 | 李浩东,闵祥玉,周雅,等.基于GBLUP等模型对梅花鹿(Cervus Nippon)生长相关性状基因组选择的预测准确性比较[J].畜牧兽医学报,2023,54(2):608-616. |
| LIH D,MINX Y,ZHOUY,et al.Comparison of prediction accuracy of genomic selection for growth-related traits in sika deer (Cervus Nippon) based on GBLUP and other models[J].Acta Veterinaria et Zootechnica Sinica,2023,54(2):608-616. | |
| 24 |
ZHANGR R,DONGY M,XINGX M.Comprehensive transcriptome analysis of sika deer antler using PacBio and Illumina sequencing[J].Sci Rep,2022,12(1):16161.
doi: 10.1038/s41598-022-20244-1 |
| 25 | 张春兰. 小尾寒羊和杜泊羊臂二头肌转录组及肌球蛋白轻链基因家族结构特征分析[D]. 泰安: 山东农业大学, 2014. |
| ZHANG C L. Transcriptome analysis of small-tailed Han sheep and Dorper's biceps Brachii and structure characteristics of myosin light chain gene families[D]. Taian: Shandong Agricultural University, 2014. (in Chinese) | |
| 26 |
MYRZABEKOVAM O,LABEITS B,NIYAZOVAR Y.Features of miRNAs binding sites within the C2H2 ZNF family: a Bos taurus, Eqqus caballus, and Ovies aries comparative approach[J].Int J Biol Chem,2020,13(1):33-46.
doi: 10.26577/ijbch.2020.v13.i1.04 |
| 27 | 林洁. 肝脏ZBTB20对胆固醇代谢的调节作用及其机制研究[D]. 上海: 中国人民解放军海军军医大学, 2023. |
| LIN J. Study on the regulation of cholesterol metabolism by liver ZBTB20[D]. Shanghai: Naval Medical University, 2023. (in Chinese) | |
| 28 | 李枫梅. 肉用牛bHLH转录因子家族成员的鉴定与分析[D]. 阜阳: 阜阳师范学院, 2017. |
| LI F M. Identification and functional analysis of the basic helix-loop-helix (bHLH) transcription factors in the meat cattle[D]. Fuyang: Fuyang Normal University, 2017. (in Chinese) | |
| 29 | 魏振宇. 山羊bHLH和PLAG基因家族分析及其与生长相关遗传标记挖掘[D]. 杨凌: 西北农林科技大学, 2022. |
| WEI Z Y. Analysis of goat bHLH and PLAG gene families and mining of genetic markers associated with growth[D]. Yangling: Northwest A&F University, 2022. (in Chinese) | |
| 30 | 鲍晶晶,浦亚斌,马月辉,等.绵羊不同发育阶段背最长肌组织中可变剪接的鉴定与分析[J].生物技术通报,2019,35(7):32-38. |
| BAOJ J,PUY B,MAY H,et al.Identification and analysis of alternative splicing in Longissimus dorsi of sheep at different development stages[J].Biotechnology Bulletin,2019,35(7):32-38. | |
| 31 | 吴倩雯,王露,王文佳,等.不同生长阶段伊拉兔骨骼肌转录组数据的挖掘分析[J].基因组学与应用生物学,2024,43(6):1028-1038. |
| WUQ W,WANGL,WANGW J,et al.Mining analysis of skeletal muscle transcriptome data of Ira rabbits at different growth stages[J].Genomics and Applied Biology,2024,43(6):1028-1038. | |
| 32 | 车隆,叶琳,吴鹏飞,等.京海黄鸡生长早期骨骼肌可变剪切分析[J].中国家禽,2022,44(12):21-27. |
| CHEL,YEL,WUP F,et al.Alternative splicing analysis of skeletal muscle for Jinghai Yellow chicken during early growth[J].China Poultry,2022,44(12):21-27. | |
| 33 |
周明帅,温晓艳,张艳,等.COL1A1基因在贵州黑山羊性腺轴中的表达及其对产羔相关基因的影响[J].农业生物技术学报,2022,30(11):2152-2162.
doi: 10.3969/j.issn.1674-7968.2022.11.009 |
|
ZHOUM S,WENX Y,ZHANGY,et al.Expression of COL1A1 gene in the gonad axis of Guizhou black goat (Capra hircus) and its effect on lambing related genes[J].Journal of Agricultural Biotechnology,2022,30(11):2152-2162.
doi: 10.3969/j.issn.1674-7968.2022.11.009 |
|
| 34 |
LIS J,MEIL Y,HEC F,et al.Identification of a family with van der Hoeve's syndrome harboring a novel COL1A1 mutation and generation of patient-derived iPSC lines and CRISPR/Cas9-corrected isogenic iPSCs[J].Human Cell,2024,37(3):817-831.
doi: 10.1007/s13577-024-01028-3 |
| 35 |
KRONENBERGH M.Developmental regulation of the growth plate[J].Nature,2003,423(6937):332-336.
doi: 10.1038/nature01657 |
| 36 |
周琳,崔磊,邓敦.母猪肢蹄病的病因、危害与预防措施[J].猪业科学,2023,40(9):61-62.
doi: 10.3969/j.issn.1673-5358.2023.09.016 |
|
ZHOUL,CUIL,DENGD.Causes, hazards and preventive measures of hoof and limb disease in sows[J].Swine Industry Science,2023,40(9):61-62.
doi: 10.3969/j.issn.1673-5358.2023.09.016 |
|
| 37 | VAN DER EEMSK L,BROWNR D,GUNDBERGC M.Circulating levels of 1, 25 dihydroxyvitamin D, alkaline phosphatase, hydroxyproline, and osteocalcin associated with antler growth in white-tailed deer[J].Acta Endocrinol (Copenh),1988,118(3):407-414. |
| 38 | 张然然,荣敏,董依萌,等.不同生长时期梅花鹿鹿茸代谢组分析[J].畜牧兽医学报,2022,53(12):4518-4526. |
| ZHANGR R,RONGM,DONGY M,et al.Metabolomic analysis of sika deer antler in different growth stages[J].Acta Veterinaria et Zootechnica Sinica,2022,53(12):4518-4526. | |
| 39 |
HUP F,WANGT J,LIUH M,et al.Full-length transcriptome and microRNA sequencing reveal the specific gene-regulation network of velvet antler in sika deer with extremely different velvet antler weight[J].Mol Genet Genomics,2019,294(2):431-443.
doi: 10.1007/s00438-018-1520-8 |
| 40 |
ZHAOY Y,LIUW M,ZHAOX M,et al.Low-density lipoprotein receptor-related protein 2 (LRP2) is required for lipid export in the midgut of the migratory locust, Locusta migratoria[J].J Integr Agric,2024,23(5):1618-1633.
doi: 10.1016/j.jia.2023.07.027 |
| 41 | 吉琳,杨秋月,方斐旻,等.鸡Apob基因组织表达与CRISPR/Cas9敲除系统的构建[J].畜牧兽医学报,2021,52(3):630-640. |
| JIL,YANGQ Y,FANGF M,et al.Tissue expression and construction of CRISPR/Cas9 knockout system of Apob in chicken[J].Acta Veterinaria et Zootechnica Sinica,2021,52(3):630-640. | |
| 42 | 刘文媛. 基于蛋白质组学和代谢组学的鹿茸区段划分与生物活性物质的分析与鉴别[D]. 北京: 中国农业科学院, 2020. |
| LIU W Y. Analysis and identification of bioactive compounds from four portions of velvet antlers in sika deer (Cervus nippon) based on proteomics and metabolomics[D]. Beijing: Chinese Academy of Agricultural Sciences, 2020. (in Chinese) | |
| 43 |
CHENY Q,POTTANATT G,SIEGELR W,et al.Angiopoietin-like protein 4 (ANGPTL4) is an inhibitor of endothelial lipase (EL) while the ANGPTL4/8 complex has reduced EL-inhibitory activity[J].Heliyon,2021,7(9):e07898.
doi: 10.1016/j.heliyon.2021.e07898 |
| 44 |
ZHANGZ S,XUL,QIUX,et al.Fibroblast growth factor 21 (FGF21) attenuates tacrolimus-induced hepatic lipid accumulation through transcription factor EB (TFEB)-regulated lipophagy[J].J Zhejiang Univ-Sci B,2023,24(6):485-495.
doi: 10.1631/jzus.B2200562 |
| 45 | 谢珂,郑卫红,谭潇.KLKs促进肿瘤增殖、迁移与侵袭的分子机制[J].中国生物化学与分子生物学报,2018,34(9):935-941. |
| XIEK,ZHENGW H,TANX.Molecular mechanism of Kallikrein-related peptidases in the promotion of tumor proliferation, migration and invasion[J].Chinese Journal of Biochemistry and Molecular Biology,2018,34(9):935-941. | |
| 46 |
DOLPHINA C,LEEA.Presynaptic calcium channels: specialized control of synaptic neurotransmitter release[J].Nat Rev Neurosci,2020,21(4):213-229.
doi: 10.1038/s41583-020-0278-2 |
| 47 | 周雅,张禾垟,刘琳玲,等.梅花鹿成纤维细胞因子受体2基因多态性及其与茸重性状的关联分析[J].中国畜牧兽医,2022,49(8):3006-3014. |
| ZHOUY,ZHANGH Y,LIUL L,et al.Analysis of FGFR2 gene polymorphism and their correlation with antler weight traits in sika deer[J].China Animal Husbandry & Veterinary Medicine,2022,49(8):3006-3014. | |
| 48 |
COURTNEYN A,BAOH,BRIGUGLIOJ S,et al.Synaptotagmin 1 clamps synaptic vesicle fusion in mammalian neurons independent of complexin[J].Nat Commun,2019,10(1):4076.
doi: 10.1038/s41467-019-12015-w |
| 49 |
ZHUY F,ZHANGX,WANGL,et al.Loss of SH3GL2 promotes the migration and invasion behaviours of glioblastoma cells through activating the STAT3/MMP2 signalling[J].J Cell Mol Med,2017,21(11):2685-2694.
doi: 10.1111/jcmm.13184 |
| [1] | 曹晋康, 张纯, 王佳瑶, 李晓彤, 王鹏宇, 方颖妍, 张昱, 丁宁, 姜力. 中国荷斯坦公牛不同耐冻性精子的蛋白质组学分析[J]. 畜牧兽医学报, 2024, 55(3): 1052-1061. |
| [2] | 王琳, 马黎, 张博, 邓俊, 张浩, 欧阳晓芳, 严达伟, 董新星. 大型迪庆藏猪不同生长阶段背脂与腹脂脂质代谢差异基因及调控网络分析[J]. 畜牧兽医学报, 2023, 54(2): 520-533. |
| [3] | 杨洋, 付宝权. 动物寄生性蠕虫基因组学研究进展[J]. 畜牧兽医学报, 2017, 48(6): 979-989. |
| 阅读次数 | ||||||
|
全文 |
|
|||||
|
摘要 |
|
|||||